PART 1 AT http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html
1 TEGOBUVIR
2 DANOPREVIR
3 CILUPREVIR
4 SOVAPREVIR
5 VEDROPREVIR
6 VANIPREVIR
7 NARLAPREVIR
8 DELDEPREVIR, NECEPREVIR
9 FALDAPREVIR
10 LEDIPASVIR
11 DACLATASVIR
12 DELEOBUVIR
13 FILIBUVIR
14 FAVIPIRAVIR
15 ASUNAPREVIR
SEE AT http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html PART 1
PART 2
16 LANINAMIVIR
17 SOFOSBUVIR
18 ELBASVIR, MK8742
19 GRAZOPREVIR MK 5172
20 BECLABUVIR, BMS-791325
21 LOPINAVIR
22 DASABUVIR, ABT 333
23 IDX 18719; IDX 719; Samatasvir
24 PALINAVIR
25 AMPRENAVIR
26
27

16 LANINAMIVIR
16 LANINAMIVIR

 472.53254, C21H36N4O8, cas no 203120-46-1, R-125489, CS-8958
472.53254, C21H36N4O8, cas no 203120-46-1, R-125489, CS-8958

Laninamivir octanoate













.........
17 SOFOSBUVIR

Sofosbuvir
 approved by the FDA")









.........................................
Synthesis of diastereomerically pure nucleotide phosphoramidates.

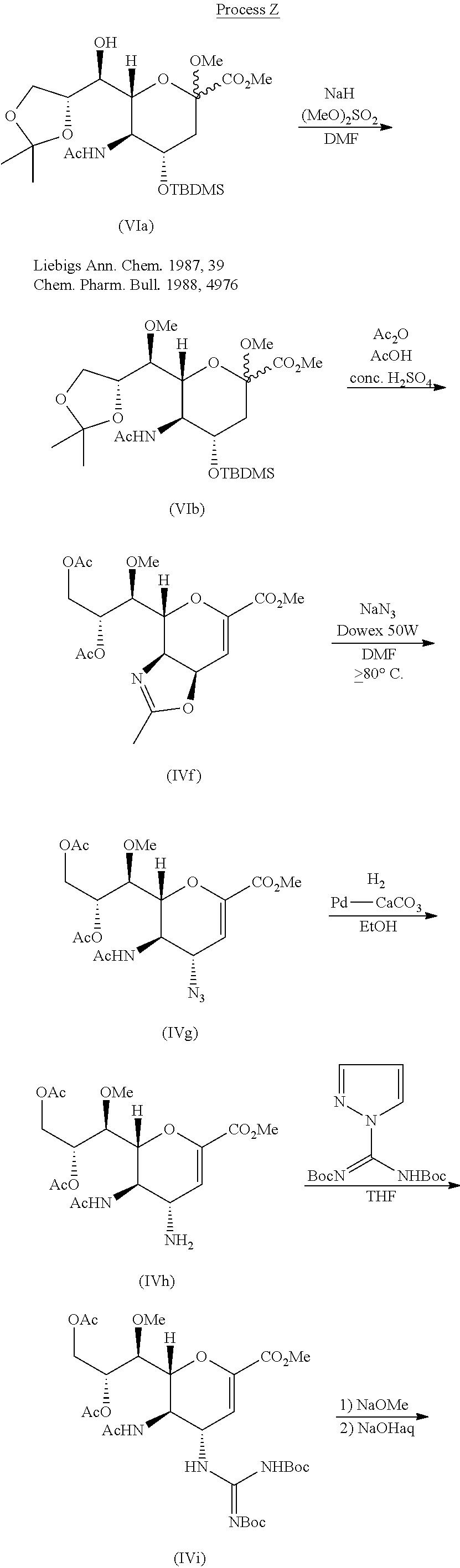
ELBASVIR, MK4732



 ELBASVIR
ELBASVIR

































































 SEEMS LIKE AN ERROR, benzene ring
SEEMS LIKE AN ERROR, benzene ring







![[(3S)-oxolan-3-yl] N-[(2S,3R)-4-[(4-aminophenyl)sulfonyl-(2-methylpropyl)amino]-3-hydroxy-1-phenylbutan-2-yl]carbamate NMR spectra analysis, Chemical CAS NO. 161814-49-9 NMR spectral analysis, [(3S)-oxolan-3-yl] N-[(2S,3R)-4-[(4-aminophenyl)sulfonyl-(2-methylpropyl)amino]-3-hydroxy-1-phenylbutan-2-yl]carbamate H-NMR spectrum](https://lh3.googleusercontent.com/blogger_img_proxy/AEn0k_tB9r0pWb-T7RpPEFsiB98Dmij5cSFNKB7uJYkagLLWpHvaQ4SOYQVCfu0mGnfduLop3qJEfA3T2fanwzvHZ24buzp2Fn_qc0IHpJ_cVkGhOVrBOX556_4R1yoBhUiSU_0bg6KsLsDPQr_EbRMO0yPc-1w=s0-d)
![[(3S)-oxolan-3-yl] N-[(2S,3R)-4-[(4-aminophenyl)sulfonyl-(2-methylpropyl)amino]-3-hydroxy-1-phenylbutan-2-yl]carbamate NMR spectra analysis, Chemical CAS NO. 161814-49-9 NMR spectral analysis, [(3S)-oxolan-3-yl] N-[(2S,3R)-4-[(4-aminophenyl)sulfonyl-(2-methylpropyl)amino]-3-hydroxy-1-phenylbutan-2-yl]carbamate C-NMR spectrum](https://lh3.googleusercontent.com/blogger_img_proxy/AEn0k_sJoHDYm6PD6kfIekANK1p5SmEi3hUA5qQOhuwMlSpED20eIiMCqkNfhtQWRxiLsutsV7EASL2Qw256ZWn3DtNEAfmyT7Ip4NNRY9STBWh-Xe0vOBSpGsdMcGihpqITtVqULmx27k0BelRF-_9s3bqqHxZ6=s0-d)

1 TEGOBUVIR
2 DANOPREVIR
3 CILUPREVIR
4 SOVAPREVIR
5 VEDROPREVIR
6 VANIPREVIR
7 NARLAPREVIR
8 DELDEPREVIR, NECEPREVIR
9 FALDAPREVIR
10 LEDIPASVIR
11 DACLATASVIR
12 DELEOBUVIR
13 FILIBUVIR
14 FAVIPIRAVIR
15 ASUNAPREVIR
SEE AT http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html PART 1
PART 2
16 LANINAMIVIR
17 SOFOSBUVIR
18 ELBASVIR, MK8742
19 GRAZOPREVIR MK 5172
20 BECLABUVIR, BMS-791325
21 LOPINAVIR
22 DASABUVIR, ABT 333
23 IDX 18719; IDX 719; Samatasvir
24 PALINAVIR
25 AMPRENAVIR
26
27
16 LANINAMIVIR
Laninamivir
(4S,5R,6R)-5-acetamido-4-carbamimidamido-6-[(1R,2R)-3-hydroxy-2-methoxypropyl]-5,6-dihydro-4H-pyran-2-carboxylic acid
| Formula | C13H22N4O7 |
|---|---|
| Mol. mass | 346.33638 g/mol |
cas 203120-17-6,
Laninamivir (L174000) prodrug; a novel long-acting neuraminidase inhibitor.
laninamivir octanoate
472.53254, C21H36N4O8, cas no 203120-46-1, R-125489, CS-8958
Daiichi Sankyo (Originator)
R-118958 is a potent, long-acting neuraminidase inhibitor (LANI) approved and launched in 2010 in Japan as an inhalable formulation for the treatment of influenza A and influenza B in adults and pediatric patients. In 2013 the product was approved in Japan for the prevention of influenza A and influenza B.
| 5-(Acetylamino)-4-[(aminoiminomethyl)amino]-2,6-anhydro-3,4,5-trideoxy-7-O-methyl-D-glycero-D-galacto-non-2-enonic Acid 9-Octanoate |
| (2R,3R,4S)-3-Acetamido-4-guanidino-2-[(1R,2R)-2-hydroxy-1-methoxy-3-(octanoyloxy)propyl]-3,4-dihydro-2H-pyran-6-carboxylic Acid |
| (4S,5R,6R)-5-Acetamido-4-guanidino-6-[(1R,2R)-2-hydroxy-1-methoxy-3-(octanoyloxy)propyl]-5,6-dihydro-4H-pyran-2-carboxylic Acid |
| CS 8958 |
ATLANTA, Dec. 20, 2013 (GLOBE NEWSWIRE) -- Biota Pharmaceuticals, Inc.
(Nasdaq:BOTA) ("Biota" or the "Company") today reported that Daiichi Sankyo Company, Limited ("Daiichi Sankyo") has been granted regulatory approval in Japan to manufacture and market Inavir(R) Dry Powder Inhaler 20mg (generic name laninamivir octanoate) for the prevention of influenza A and B. Inavir(R) was successfully developed and launched by Daiichi Sankyo in Japan for treatment of influenza A and B viruses in October, 2010. Biota is developing laninamivir octanoate outside of Japan for the treatment of influenza, and is currently conducting a large, multi-national Phase 2 trial of laninamivir octanoate in adults infected with influenza. In 2003, the Company and Daiichi Sankyo entered into a collaboration and license agreement to develop long-acting neuraminidase inhibitors, including laninamivir octanoate, and in March 2009, the parties entered into a commercialization agreement, pursuant to which Daiichi Sankyo obtained exclusive marketing rights to laninamivir octanoate in Japan.http://www.pharmalive.com/biota-flu-drug-okd-in-japan
(Nasdaq:BOTA) ("Biota" or the "Company") today reported that Daiichi Sankyo Company, Limited ("Daiichi Sankyo") has been granted regulatory approval in Japan to manufacture and market Inavir(R) Dry Powder Inhaler 20mg (generic name laninamivir octanoate) for the prevention of influenza A and B. Inavir(R) was successfully developed and launched by Daiichi Sankyo in Japan for treatment of influenza A and B viruses in October, 2010. Biota is developing laninamivir octanoate outside of Japan for the treatment of influenza, and is currently conducting a large, multi-national Phase 2 trial of laninamivir octanoate in adults infected with influenza. In 2003, the Company and Daiichi Sankyo entered into a collaboration and license agreement to develop long-acting neuraminidase inhibitors, including laninamivir octanoate, and in March 2009, the parties entered into a commercialization agreement, pursuant to which Daiichi Sankyo obtained exclusive marketing rights to laninamivir octanoate in Japan.http://www.pharmalive.com/biota-flu-drug-okd-in-japan
Laninamivir (CS-8958) is a neuraminidase inhibitor which is being researched for the treatment and prophylaxis of Influenzavirus A and Influenzavirus B.[1] It is currently in Phase III clinical trials. [2]
Laninamivir was approved for influenza treatment in Japan in 2010 and is currently marketed under the name "Inavir" by Daiichi Sankyo. Biota Pharmaceuticals [3] and Daiichi Sankyo co-own Laninamivir. On 1st April 2011, BARDA awarded up to an estimated U$231m to Biota Pharmaceuticals (Formerly Biota Holdings Ltd) wholly owned subsidiary, Biota Scientific Management Pty Ltd, for the advanced development of Laninamivir in the US. [4]
patent
| US2010204314 | 8-13-2010 | DRUG FOR TREATMENT OF INFLUENZA |
WO 2013089168
WO 2008126943
The recent flu scares – first H5N1 bird flu and then H1N1 swine flu – transformed Roche’s neuraminidase inhibitor Tamiflu (oseltamivir) into a household name, along with GSK’s Relenza (zanamivir). Both of these require twice-daily dosing, and the orally available oseltamivir is the first choice, but resistance is starting to appear.
A new neuraminidase inhibitor, laninamivir, is being developed by Daiichi Sankyo.5 When administered as the octanoate prodrug form, it appears that a single dose might be sufficient to treat influenza, weekly doses could be preventative, and it is active against extremely pathogenic H5N1 strains.
Laninamivir octanoate
In a double blind, randomised, placebo-controlled Phase I study in 76 healthy male volunteers, subjects were given inhaled single doses of 5, 10, 20, 40, 80 or 120mg of the prodrug, or twice-daily doses of 20 or 40mg for three days.6 No adverse events were observed, and while the prodrug disappeared from the plasma with a half-life of about two hours, the laninamivir itself was much more slowly eliminated, with a half-life of the order of three days, suggesting the potential for giving long-lasting activity against influenza.
In another Phase I trial, a total of 20 healthy subjects with renal function ranging from normal to severely impaired were given single inhaled 20mg doses of the prodrug.7 The degree of renal impairment did not affect the maximum concentration or the time to achieve it, but the half-life increased as renal function reduced. This indicates that the rate-limiting step for elimination is drug release rate to plasma from tissues rather than renal excretion. It was well tolerated, but systemic exposure increased with increasing renal impairment.
It has also been compared with oseltamivir in patients with influenza. A total of 186 children under 10 who had had febrile influenza symptoms for no longer than 36 hours were randomised to receive 20 or 40mg of laninamivir octanoate as a single inhalation or 2mg/kg oseltamivir orally twice a day for five days.8
The new drug gave a significant reduction, of 61 hours for the 40mg group and 66 for the 20mg group, in median time to illness alleviation compared with oseltamivir in those with oseltamivir-resistant H1N1 influenza A. However, there was no significant difference in the time to alleviation of illness with H3N2 influenza A, or influenza B.
The most common side-effects were gastrointestinal problems.
In a Phase III trial, a total of 1,003 adult patients with febrile influenza symptoms for no more than 36 hours were given similar doses to those in the trial in children.9 Median time to alleviation of illness was 73h for 40mg, 86h for 20mg, and 74h for oseltamivir, and the proportion of patients shedding virus at day 3 was significantly lower in the 40mg group than for those given oseltamivir.
- Yamashita M, Tomozawa T, Kakuta M, Tokumitsu A, Nasu H, Kubo S (January 2009)."CS-8958, a prodrug of the new neuraminidase inhibitor R-125489, shows long-acting anti-influenza virus activity". Antimicrobial Agents and Chemotherapy 53 (1): 186–92.doi:10.1128/AAC.00333-08. PMC 2612152. PMID 18955520.
- Hayden F (January 2009). "Developing new antiviral agents for influenza treatment: what does the future hold?". Clinical Infectious Diseases. 48. Suppl 1 (S1): S3–13.doi:10.1086/591851. PMID 19067613.
- http://www.biotapharma.com
- http://www.biotapharma.com/?page=1021001&subpage=1021019
5. T. Honda et al. Synthesis and in vivo influenza virus-inhibitory effect of ester prodrug of 4-guanidino-7-O-methyl-Neu5Ac2en, Bioorg Med Chem Lett 2009, 19(11): 2938
6. H. Ishizuka et al. J. Clin. Pharmacol. 2010, 50, 1319
7. H. Ishizuka et al. J. Clin. Pharmacol. 2010, epub ahead of print, doi 10.1177/0091270010361914
8. N. Sugaya and Y. Ohashi, Antimicrob. Ag. Chemother. 2010, 54, 2575
9 A. Watanabe et al. Clin. Inf. Dis. 2010, 51, 1167
A new route toward 2-acetamido-4-O-methyl-2-deoxy-D-mannopyranose from a Ferrier derivative of tri-O-acetyl-D-glucal, which contributes to aldolase-catalyzed synthesis of laninamivir (CS-8958)
Tetrahedron 2013, 39(37): 7931
Tetrahedron 2013, 39(37): 7931
Infection of poultry with H5N1 avian influenza virus has been expanding since 2003 in wide areas including Asia, Europe and Africa. Humans infected with this virus have been found not only in Asia but also in Middle East and Africa. If a new type of H5N1 influenza virus has appeared and its infection has started, it is believed that the infection will rapidly expand to cause a worldwide spread (i.e., influenza pandemic) because most people do not possess immunity against that virus and influenza viruses spread via droplet infection and airborne infection. More than half of human patients infected with H5N1 influenza virus have died so far. Thus, the virus is highly pathogenic. It is known that three influenza pandemics, the Spanish Flu, the Asian Flu and the Hong Kong Flu, occurred in the 20th century. In the Spanish Flu which caused the largest number of patients, it is estimated that about 20-40 million people died in the world and about 0.5 million people in Japan.
According to a report from Japanese Ministry of Health, Labour and Welfare made in November, 2005, if a new type influenza virus has spread, the number of patients who will consult medical doctors in Japan as a result of infection with that virus is estimated about 18-25 million. Further, when the pathogenicity of that new type influenza virus is severe, the number of inpatients is estimated about 0.2 million while the number of dead is estimated about 0.64 million. Therefore, not only health hazard but also significant influences upon social activities are feared.
Thus, a new type influenza can cause a highly severe disease. Early development of effective therapeutics is demanded.
Although it is reported that zanamivir (in particular, zanamivir hydrate) and oseltamivir (in particular, oseltamivir phosphate or oseltamivir carboxylate) which are influenza therapeutics with neuraminidase inhibitory activity show an inhibitory activity against H5N1 influenza virus, compounds with more excellent activity are desired (Non-Patent Document 1 or 2). Further, H5N1 influenza virus strains against which oseltamivir does not show any inhibitory activity (i.e., oseltamivir resistant virus strains) have been reported. Compounds which possess an inhibitory activity against these oseltamivir resistant H5N1 influenza virus strains are desired (Non-Patent Document 1 or 2).
Compounds represented by formula (I) are known to be useful as influenza therapeutics with neuraminidase inhibitory activity (Patent Documents 1 to 3). However, it has not been reported that these compounds have an inhibitory activity against H5N1 influenza virus. Further, the structures of the compounds represented by formula (I) resemble the structure of zanamivir but are completely different from the structure of oseltamivir.
Non-Patent Document 1: Nature, 2005, vol. 437, p. 1108
Non-Patent Document 2: N. Engl. J. Med., 2005, vol. 353, (25):2667-72
Patent Document 1: U.S. Pat. No. 6,340,702 (Japanese Patent No. 3209946)
Patent Document 2: U.S. Pat. No. 6,451,766 (Japanese Patent Publication No. Hei 10-109981)
Patent Document 3: U.S. Pat. No. 6,844,363 (Japanese Patent Publication No. 2002-012590)
Patent Document 1: U.S. Pat. No. 6,340,702 (Japanese Patent No. 3209946)
Patent Document 2: U.S. Pat. No. 6,451,766 (Japanese Patent Publication No. Hei 10-109981)
Patent Document 3: U.S. Pat. No. 6,844,363 (Japanese Patent Publication No. 2002-012590)
...........................
Preparation Example 1 5-Acetamido-4-guanidino-9-O-octanoyl-2,3,4,5-tetradeoxy-7-O-methyl-D-glycero-D-galacto-non-2-enopyranosoic acid
(1) Diphenylmethyl 5-acetamido-4-(N,N-bis-t-butyloxycarbonyl)guanidino-9-O-octanoyl-2,3,4,5-tetradeoxy-7-O-methyl-D-glycero-D-galacto-non-2-enopyranosoate (3.46 g, 4.1 mmol) disclosed in Example 35 (i) of U.S. Pat. No. 6,340,702 (Japanese Patent No. 3209946) was dissolved in methylene chloride (27 ml) and trifluoroacetic acid (14 ml). The resultant solution was stirred at room temperature overnight. The reaction solution was concentrated to dryness under reduced pressure, followed by three cycles of azeotropic distillation to dryness with toluene (5 ml). The resultant oily material was dissolved in ethyl acetate (10 ml). The solution was poured into a saturated aqueous solution of sodium hydrogencarbonate (50 ml). The pH of the resultant solution was adjusted to 8.5 by addition of 20% aqueous solution of sodium carbonate. Then, the solution was stirred at room temperature for 3 hr and its pH was adjusted to 5.0 with hydrochloric acid (3 ml), followed by stirring at room temperature for another 1 hr. The solution was further stirred for 1 hr while ice-cooling. Subsequently, precipitating crystals were suction filtered and vacuum dried for 10 hr at an external temperature of 50° C. The resultant crystals were left in the air for one day to thereby yield the subject compound as a hydrate crystal (0.97 g; yield 51%).
Infrared Absorption Spectrum (KBr) ν max cm−1: 3412, 2929, 2856, 1676, 1401, 1320, 1285, 1205, 1137, 722.
1H Nuclear Magnetic Resonance Spectrum (400 MHz, CD3OD) δ ppm: 5.88 (1H, d, J=2.5 Hz), 4.45 (3H, m), 4.27 (1H, dd, J=10.0 Hz, 10.0 Hz), 4.15 (1H, m), 3.47 (21-1, m), 3.42 (3H, s), 2.37 (2H, t, J=7.4 Hz), 2.10 (3H, s), 1.31 (2H, m), 1.20-1.40 (8H, m), 0.85-0.95 (3H, m).
13C Nuclear Magnetic Resonance Spectrum (100 MHz, CD3OD) δ ppm: 176.5, 173.7, 164.7, 158.9, 146.7, 108.7, 80.1, 78.0, 69.3, 66.8, 61.4, 52.4, 35.1, 32.8, 30.2, 30.1, 26.0, 23.7, 22.8, 14.4.
(2) The subject compound was also obtained by the method described below.
5-Acetamido-4-guanidino-9-O-octanoyl-2,3,4,5-tetradeoxy-7-O-methyl-D-glycero-D-galacto-non-2-enopyranosoic acid trifluoroacetic acid salt (3.0 g, 5.1 mmol) disclosed in Example 35 (ii) of U.S. Pat. No. 6,340,702 (Japanese Patent No. 3209946) was subjected to reversed phase column chromatography [Cosmosil 75C 18PREP (nacalai tesque), 100 g] and eluted with methanol/water (0/1-1/1-2/1). Fractions containing the compound of interest were vacuum concentrated. The precipitating crystals were suction filtered and vacuum dried. The resultant crystals were left in the air for one day to thereby yield the subject compound as a hydrate crystal (1.2 g; yield 49%). The property data of the resultant compound were consistent with those of the compound obtained in (1) above.
Preparation Example 2 5-Acetamido-4-guanidino-2,3,4,5-tetradeoxy-7-O-methyl-D-glycero-D-galacto-non-2-enopyranosoic acid
5-Acetamido-4-guanidino-2,3,4,5-tetradeoxy-7-O-methyl-D-glycero-D-galacto-non-2-enopyranosoic acid trifluoroacetic acid salt (3.0 g, 5.1 mmol) disclosed in Example 28 (viii) of U.S. Pat. No. 6,340,702 (Japanese Patent No. 3209946) was purified in an ion-exchange resin column [Dowex-50X; (i) water and (ii) 5% aqueous ammonium solution] and further purified by reversed phase column chromatography [Cosmosil 75C 18PREP (nacalai tesque); water]. Fractions containing the compound of interest were vacuum concentrated. The resultant solid was washed with methanol, filtered and dried to thereby yield the subject compound (1.43 g) as a colorless solid.
1H Nuclear Magnetic Resonance Spectrum (400 MHz, CD3OD) δ ppm: 5.64 (1H, d, J=2.0 Hz), 4.43 (2H, m), 4.22 (1H, dd, J=10.0 Hz, 10.0 Hz), 4.00 (1H, m), 3.85 (1H, dd, J=10.0 Hz, 2.4 Hz), 3.68 (1H, dd, J=10.0 Hz, 5.5 Hz), 3.58 (1H, m), 3.43 (3H, s).
...............................
................................
Process W is known as a method for manufacturing a compound represented by the formula (Ia), which is embraced in a compound represented by the formula (I) or a pharmacologically acceptable salt thereof, (hereinafter also referred to as “compound (Ia)”; the same shall be applied with respect to other (Patent Document 1). In Process W, n-Hep represents a 1-heptyl group.
Process X is known as a method for manufacturing compound (Ib), which is embraced in compound (I) or a pharmacologically acceptable salt thereof (Patent Document 2). Compound (IVk) is a synthetic intermediate in Process W. In Process X, n-Hep represents a 1-heptyl group.
Process Y is known as a method for manufacturing compound (IIIa), which is a trifluoroacetic acid salt of compound (III) (Non-patent Document 1). The procedures from compound (IVc) to compound (IVe) and from compound (IVf) to compound (IVh) in Process Y are the same as in Process W.
Process Z is known as a method for manufacturing compound (IIIa), which is a trifluoroacetic acid salt of compound (III) (Non-patent Document 2). In Process Z, the procedure from compound (IVf) to compound (IVh) is the same as in Process W, and the procedure from compound (IVh) to compound (IIIa) is the same as in Process Y.
From the viewpoint of industrial production, the aforementioned Process W, Process Y, or Process Z could be improved in points such as the following:
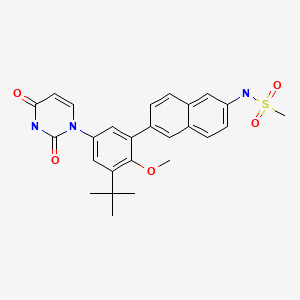
Sofosbuvir
Sovaldi
M.Wt: 529.45
Formula: C22H29FN3O9P
Isopropyl (2S)-2-[[[(2R,3R,4R,5R)-5-(2,4-dioxopyrimidin-1-yl)-4-fluoro-3-hydroxy-4-methyl-tetrahydrofuran-2-yl]methoxy-phenoxy-phosphoryl]amino]propanoate
A prodrug of 2'-deoxy-2'-alpha-F-2'-beta-C-methyluridine 5'-monophosphate.
GS-7977, PSI-7977
GS-7977, PSI-7977
- GS 7977
- GS-7977
- PSI 7977
- PSI-7977
- Sofosbuvir
- Sovaldi
- UNII-WJ6CA3ZU8B
CAS Registry Number :1190307 -88-0
Indications: Chronic hepatitis C (HCV GT1, GT2, GT3, GT4)
Mechanism: nucleoside NS5B polymerase inhibitor
approved Time: December 6, 2013
,U.S. Patent Number: 7964580,8415322,8334270,7429572;, patent validity: March 26, 2029 (U.S. Patent No.: 7,964,580 and 8,334,270), April 3, 2025 (U.S. Patent No.: 7,429,572 and 8,415,322)
Mechanism: nucleoside NS5B polymerase inhibitor
approved Time: December 6, 2013
,U.S. Patent Number: 7964580,8415322,8334270,7429572;, patent validity: March 26, 2029 (U.S. Patent No.: 7,964,580 and 8,334,270), April 3, 2025 (U.S. Patent No.: 7,429,572 and 8,415,322)
US patent number 7964580, US patent number 8415322, US patent number 8334270,US patent number 7429572 Patent Expiration Date: March 26, 2029 for US patent number 7964580 and 8334270 (2028 in EU); April 3, 2025 for US patent number 7429572 and 8415322
Sales value (estimated): $ 1.9 billion (2014), 6600000000 USD (2016)
Drug Companies: Gilead Sciences, Inc. (Gilead Sciences)
WASHINGTON, Dec. 6, 2013 (AP) — Federal health officials have approved a highly anticipated hepatitis C drug from Gilead Sciences Inc. that is expected to offer a faster, more palatable cure to millions of people infected with the liver-destroying virus.
The Food and Drug Administration said Friday it approved the pill Sovaldi in combination with older drugs to treat the main forms of hepatitis C that affect U.S. patients.
Current treatments for hepatitis C can take up to a year of therapy and involve weekly injections of a drug that causes flu-like side effects. That approach only cures about three out of four patients. Sovaldi is a daily pill that in clinical trials cured roughly 90 percent of patients in just 12 weeks, when combined with the older drug cocktail.http://www.pharmalive.com/us-approves-breakthrough-hepatitis-c-drug
- The end of October 2013 saw a nod from the FDA given to Gilead’s New Drug Application for Sofosbuvir, a much needed treatment for hepatitis C.
- As a nucleotide analogue, Sofosbuvir is designed as a once daily treatment.
- There are roughly 170 million cases of hepatitis C around the world.
- A report in the Journal of the American Medical Association on August 28, 2013 revealed that the Sofosbuvir and Ribavirin combination treatment effectively cured many patients with the Hepatitis C Virus.
- The Sofosbuvir and Ribavirin drug combination was void of interferon-based treatments, which many patients are resistant too.
- More than 3 million Americans have chronic Hepatitis C Virus, and 22 percent of these patients are African American.
Sofosbuvir (brand names Sovaldi and Virunon) is a drug used for hepatitis C virus (HCV) infection, with a high cure rate.[1][2] It inhibits the RNA polymerase that the hepatitis C virus uses to replicate its RNA. It was discovered at Pharmasset and developed by Gilead Sciences.[3]
Sofosbuvir is a component of the first all-oral, interferon-free regimen approved for treating chronic Hepatitis C.[4]
In 2013, the FDA approved sofosbuvir in combination with ribavirin (RBV) for oral dual therapy of HCV genotypes 2 and 3, and for triple therapy with injected pegylated interferon (pegIFN) and RBV for treatment-naive patients with HCV genotypes 1 and 4.[4] Sofosbuvir treatment regimens last 12 weeks for genotypes 1, 2 and 4, compared to 24 weeks for treatment of genotype 3. The label furhter states that sofosbuvir in combination with ribavirin may be considered for patients infected with genotype 1 who are interferon-ineligible.[5] Sofosbuvir will cost $84,000 for 12 weeks of treatment and $168,000 for the 24 weeks, which some patient advocates have criticized as unaffordable.
Interferon-free therapy for treatment of hepatitis C eliminates the substantial side-effects associated with use of interferon. Up to half of hepatitis C patients cannot tolerate the use of interferon.[6]
Sofosbuvir is a prodrug that is metabolized to the active antiviral agent 2'-deoxy-2'-α-fluoro-β-C-methyluridine-5'-triphosphate.[7] Sofosbuvir is anucleotide analog inhibitor of the hepatitis C virus (HCV) polymerase.[8] The HCV polymerase or NS5B protein is a RNA-dependent RNA polymerase critical for the viral cycle.
The New Drug Application for Sofosbuvir was submitted on April 8, 2013 and received the FDA's Breakthrough Therapy Designation, which grants priority review status to drug candidates that may offer major treatment advantages over existing options.[9]
On 6th December 2013, the U.S. Food and Drug Administration approved sofosbuvir for the treatment of chronic hepatitis C.[10]
Sofosbuvir is being studied in combination with pegylated interferon and ribavirin, with ribavirin alone, and with other direct-acting antiviral agents.[11][12] It has shown clinical efficacy when used either with pegylated interferon/ribavirin or in interferon-free combinations. In particular, combinations of sofosbuvir with NS5A inhibitors, such as daclatasvir or GS-5885, have shown sustained virological response rates of up to 100% in people infected with HCV.[13]
Data from the ELECTRON trial showed that a dual interferon-free regimen of sofosbuvir plus ribavirin produced a 24-week post-treatment sustained virological response (SVR24) rate of 100% for previously untreated patients with HCV genotypes 2 or 3.[14][15]
Data presented at the 20th Conference on Retroviruses and Opportunistic Infections in March 2013 showed that a triple regimen of sofosbuvir, ledipasvir, and ribavirin produced a 12-week post-treatment sustained virological response (SVR12) rate of 100% for both treatment-naive patients and prior non-responders with HCV genotype 1.[16] Gilead has developed a sofosbuvir + ledipasvir coformulation that is being tested with and without ribavirin.
Sofosbuvir will cost $84,000 for 12 weeks of treatment used for genotype 1 and 2, and $168,000 for the 24 weeks used for genotype 3.[17] This represents a substantial pricing increase from previous treatments consisting of interferon and ribavirin, which cost between $15,000 and $20,000.[18] The price is also significantly higher than that of Johnson & Johnson's recently approved drug simeprevir (Olysio), which costs $50,000 and also treats chronic hepatitis C.[18] The high cost of the drug has resulted in a push back from insurance companies and the like, includingExpress Scripts, which has threatened to substitute lower priced competitors, even if those therapies come with a more unfriendly dosing schedule.[18] Other treatments that have recently entered the market have not matched the efficacy of sofosbuvir, however, allowing Gilead to set a higher price until additional competition enters the market.[18] Patient advocates such as Doctors Without Borders have complained about the price, which is particularly difficult for underdeveloped countries to afford.[19]

sofosbuvir
- News: United States to approve potent oral drugs for hepatitis C, Sara Reardon, Nature, 30 October 2013
- Sofia MJ, Bao D, Chang W, Du J, Nagarathnam D, Rachakonda S, Reddy PG, Ross BS, Wang P, Zhang HR, Bansal S, Espiritu C, Keilman M, Lam AM, Steuer HM, Niu C, Otto MJ, Furman PA (October 2010). "Discovery of a β-d-2'-deoxy-2'-α-fluoro-2'-β-C-methyluridine nucleotide prodrug (PSI-7977) for the treatment of hepatitis C virus". J. Med. Chem. 53 (19): 7202–18.doi:10.1021/jm100863x. PMID 20845908.
- "PSI-7977". Gilead Sciences.
- Tucker M (December 6, 2013). "FDA Approves 'Game Changer' Hepatitis C Drug Sofosbuvir". Medscape.
- "U.S. Food and Drug Administration Approves Gilead’s Sovaldi™ (Sofosbuvir) for the Treatment of Chronic Hepatitis C - See more at: http://www.gilead.com/news/press-releases/2013/12/us-food-and-drug-administration-approves-gileads-sovaldi-sofosbuvir-for-the-treatment-of-chronic-hepatitis-c#sthash.T9uTbSWK.dpuf". Gilead. December 6, 2013.
- "Sofosbuvir is safer than interferon for hepatitis C patients, say scientists". News Medical. April 25, 2013.
- Murakami E, Tolstykh T, Bao H, Niu C, Steuer HM, Bao D, Chang W, Espiritu C, Bansal S, Lam AM, Otto MJ, Sofia MJ, Furman PA (November 2010). "Mechanism of activation of PSI-7851 and its diastereoisomer PSI-7977". J. Biol. Chem. 285 (45): 34337–47.doi:10.1074/jbc.M110.161802. PMC 2966047. PMID 20801890.
- Alejandro Soza (November 11, 2012). "Sofosbuvir". Hepaton.
- "FDA Advisory Committee Supports Approval of Gilead’s Sofosbuvir for Chronic Hepatitis C Infection". Drugs.com. October 25, 2013.
- "FDA approves Sovaldi for chronic hepatitis C". FDA New Release. U.S. Food and Drug Administration. 2013-12-06.
- Murphy T (November 21, 2011). "Gilead Sciences to buy Pharmasset for $11 billion".Bloomberg Businessweek.
- Asselah T (January 2014). "Sofosbuvir for the treatment of hepatitis C virus". Expert Opin Pharmacother 15 (1): 121–30. doi:10.1517/14656566.2014.857656. PMID 24289735.
- "AASLD 2012: Sofosbuvir and daclatasvir dual regimen cures most people with HCV genotypes 1, 2, or 3". News. European Liver Patients Association. 2012-11-21.
- AASLD: PSI-7977 plus Ribavirin Can Cure Hepatitis C in 12 Weeks without Interferon. Highleyman, L. HIVandHepatitis.com. 8 November 2011.
- Gane EJ, Stedman CA, Hyland RH, Ding X, Svarovskaia E, Symonds WT, Hindes RG, Berrey MM (January 2013). "Nucleotide polymerase inhibitor sofosbuvir plus ribavirin for hepatitis C".N. Engl. J. Med. 368 (1): 34–44. doi:10.1056/NEJMoa1208953. PMID 23281974.
- CROI 2013: Sofosbuvir + Ledipasvir + Ribavirin Combo for HCV Produces 100% Sustained Response. Highleyman, L. HIVandHepatitis.com. 4 March 2013.
- Campbell T (December 11, 2013). "Gilead's Sofosbuvir Gets New Name, Price, Headaches". The Motley Fool.
- Cohen, J. (2013). "Advocates Protest the Cost of a Hepatitis C Cure". Science 342 (6164): 1302–1303. doi:10.1126/science.342.6164.1302. PMID 24337268. edit
The chemical structure
 approved by the FDA")
GS-7977, (S)-isopropyl 2-(((S)-(((2R,3R,4R,5R)-5-(2,4-dioxo-3,4- dihydropyrimidin^l(2H)-yl)-4-fluoro-3-hydroxy-4-methyltetrahydrofuran-2- yl)methoxy)(phenoxy)phosphoryl)amino)propanoate, available from Gilead Sciences, Inc., is described and claimed in U.S. Patent No. 7,964,580. (See also US 2010/0016251, US 2010/0298257, US 201 1/0251 152 and US 2012/0107278.) GS-7977 has the structure:
GS-7977 can be crystalline or amorphous. Examples of preparing crystalline and amorphous forms of GS-7977 are disclosed in US 2010/0298257 (US 12/783,680) and US 201 1/0251 152 (US 13/076,552),
Commerically available isopropylidine protected D-glyceraldehyde was reacted with (carbethoxyethylidene)triphenylmethylphosphorane gave the chiral pentenoate ester YP-1. Permanganate dihydroxylation of YP-1 in acetone gave the D-isomer diol YP-2. The cyclic sulfate YP-3 was obtained by first making the cyclic sulfite with thionyl chloride and then oxidizing to cyclic sulfate with sodium hypochlorite. Fluorination of YP-3 with triethylamine-trihydrofluoride(TEA-3HF) in the presence of triethylamine, followed by the hydrolysis of sulfate ester in the presence of concentrated HCl provided diol YP-4 which was benzoylated to give ribonolactone YP-5. Reduction of YP-5 with Red-Al followed by chlorination with sulfuryl chloride in the presence of catalytic amount of tetrabutylammonium bromide yielded YP-6. The conversion of YP-6 to benzoyl protected 2′-deoxyl-2′-alpha-F-2′-Beta-C-methylcytidine (YP-7) was achieved by using O-trimethyl silyl-N4-benzoylcytosine and stannic chloride. Preparation of the uridine nucleoside YP-8 was accomplished by first heating benzoyl cytidine YP-7 in acetic acid then treating with methoanolic ammonia to provide YP-8 in 78% yield.
The phosphoramidating reagent YP-9 was obtained by first reacting phenyldichlorophosphate with L-Alanine isopropyl ester hydrochloride and then with pentafluorophenol. Isolation of single Sp diastereomer YP-9 was achieved via crystallization-induced dynamic resolution in the presence of 20% MTBE/hexane at room temperature.
The uridine nucleoside YP-8 was treated with tert-butylmagnesium chloride in dry THF, followed by pentafluorophenyl Sp diastereomer YP-9 to furnish the Isopropyl (2S)-2-[[[(2R,3R,4R,5R)-5-(2,4-dioxopyrimidin-1-yl)-4-fluoro-3-hydroxy-4-methyl-tetrahydrofuran-2-yl]methoxy-phenoxy-phosphoryl]amino]propanoate (Sovaldi, sofosbuvir, GS-7977, PSI-7977)。
............
US 7429572
US 8415322
US 7964580
US 8334270B
WO 2006012440
WO 2011123668
...................................................
In US 20050009737 published Jan. 13, 2005, J. Clark discloses fluoro-nucleoside derivatives that inhibit Hepatitis C Virus (HCV) NS5B polymerase. In particular, 4-amino-1-((2R,3R,4R,5R)-3-fluoro-4-hydroxy-5-hydroxymethyl-3-methyl-tetrahydro-faran-2-yl)-1H-pyrimidin-2-one (18) was a particularly potent inhibitor of HCV polymerase as well as the polymerase of other Flaviviridae.

In WO2006/012440 published Feb. 2, 2006, P. Wang et al disclose processes for the preparation of 18. Introduction of the cytosine is carried out utilizing the Vorbruggen protocol. In US 20060122146 published Jun. 8, 2006, B.-K. Chun et al. disclose and improved procedures for the preparation of the 2-methyl-2-fluoro-lactone 10. In the latter disclosure the nucleobase is glycosylated by reacting with ribofuranosyl acetate which is prepared by reduction of 10 with LiAlH(O-tert-Bu)3 followed by acetylaton of the intermediate lactol which was treated with an O-trimethylsilyl N4-benzoylcytosine in the presence of SnCl4 to afford the O,O,N-tribenzoylated nucleoside.
.........................................................................
The present process as described in SCHEME A and the following examples contain numerous improvements which have resulted in higher yields of the desired nucleoside. The asymmetric hydroxylation of 22 was discovered to be best carried out with sodium permanganate in the presence of ethylene glycol, sodium bicarbonate in acetone which afforded the diol in 60-64% on pilot plant scale. The sodium permanganate procedure avoids introduction of osmium into the process stream. Further more the stereospecific hydroxylation can be accomplished without using an expensive chiral ligand. The requisite olefin is prepared from (1S,2S)-1,2-bis-((R)-2,2-dimethyl-[1,3]dioxolan-4-yl)-ethane-1,2-diol (20) (C. R. Schmid and J. D. Bryant, Org. Syn. 1995 72:6-13) by oxidative cleavage of the diol and treating the resulting aldehyde with 2-(triphenyl-λ5-phosphanylidene)-propionic acid ethyl ester to afford 22.

(i) NaIO4, NaHCO3, DCM; (ii) MeC(═PPh3)CO2Et; (iii) acetone-NaMnO4 (aq), ethylene glycol, NaHCO3, −10 to 0° C.; aq. NaHSO3 (quench); (iv) i-PrOAc, MeCN, TEA, SOCl2; (v) i-PrOAc, MeCN, NaOCl; (vi) TEA-3HF, TEA; (vii) HCl (aq)-BaCl2-aq; (viii) (PhCO)2O, DMAP, MeCN, (ix) RED-AL/TFE (1:1), DCM; (x) SO2Cl2-TBAB, DCM; (xi) 32, SnCl4-PhCl; (xii) MeOH-MeONa
EXAMPLE 3 (2S,3R)-3-[(4R)-2,2-dimethyl-[1,3]dioxolan-4-yl]-2,3-dihydroxy-2-methyl-propionic acid ethyl ester (24)

A suspension of 22 (10 kg, CAS Reg. No. 81997-76-4), ethylene glycol (11.6 kg), solid NaHCO3 (11.8 kg) and acetone (150 L) is cooled to ca.-15° C. A solution of 36% aqueous NaMnO4 (19.5 kg) is charged slowly (over 4 h) to the suspension maintaining reaction temperature at or below −10° C. After stirring for 0.5 h at −10° C., an aliquot of the reaction mixture (ca. 5 mL) is quenched with 25% aqueous sodium bisulfite (ca. 15 mL). A portion of resulting slurry is filtered and submitted for GC analysis to check the progress of the reaction. When the reaction is complete, the reaction mixture is quenched by slow addition (over 40 min) of cooled (ca. 0° C.) 25% aqueous NaHSO3 (60 L). The temperature of the reaction mixture is allowed to reach 4° C. during the quench. CELITE® (ca. 2.5 kg) is then slurried in acetone (8 kg) and added to the dark brown reaction mixture. The resulting slurry is aged at RT to obtain light tan slurry. The slurry is filtered, and the filter cake is washed with acetone (3×39 kg). The combined filtrate is concentrated by vacuum distillation (vacuum approximately 24 inches of Hg; max pot temperature is 32° C.) to remove the acetone. The aqueous concentrate is extracted with EtOAc (3×27 kg), and the combined organic extracts were washed with water (25 L). The organic phase is then concentrated by atmospheric distillation and EtOAc is replaced with toluene. The volume of the batch is adjusted to ca. 20 L. Heptane (62 kg) is added and the batch cooled to ca. 27° C. to initiate crystallization. The batch is then cooled to −10° C. After aging overnight at −10° C., the product is filtered, washed with 10% toluene in heptane and dried at 50° C. under vacuum to afford 6.91 kg (59.5%) of 24 (CARN 81997-76-4) as a white crystalline solid.
EXAMPLE 4 (3R,4R,5R)-3-Fluoro-4-hydroxy-5-hydroxymethyl-3-methyl-dihydro-furan-2-one (10)

steps 1 & 2—A dry, clean vessel was charged with 24 (6.0 kg), isopropyl acetate (28.0 kg), MeCN (3.8 kg) and TEA (5.4 kg). The mixture was cooled to 5-10° C., and thionyl chloride (3.2 kg) was added slowly while cooling the solution to maintain the temperature below 20° C. The mixture was stirred until no starting material was left (GC analysis). The reaction was typically complete within 30 min after addition is complete. To the mixture was added water (9 kg) and after stirring, the mixture was allowed to settle. The aqueous phase was discarded and the organic phase was washed with a mixture of water (8 kg) and saturated NaHCO3 (4 kg) solution. To the remaining organic phase containing 36 was added MeCN (2.5 kg) and solid NaHCO3 (3.1 kg). The resulting slurry was cooled to ca. 10° C. Bleach (NaOCl solution, 6.89 wt % aqueous solution, 52.4 kg, 2 eq.) was added slowly while cooling to maintain temperature below 25° C. The mixture was aged with stirring over 90-120 min at 20-25° C., until the reaction was complete (GC analysis). After completion of the reaction, the mixture was cooled to ca. 10° C. and then quenched with aqueous Na2SO3 solution (15.1% w/w, 21 kg) while cooling to maintain temperature below 20° C. The quenched reaction mixture was filtered through a cartridge filter to remove inorganic solids. The filtrate was allowed to settle, and phases are separated and the aqueous phase is discarded. The organic layer was washed first with a mixture of water (11 kg) and saturated NaHCO3 solution (4.7 kg), then with of saturated NaHCO3 solution (5.1 kg). DIPEA (220 mL) was added to the organic phase and the resulting solution was filtered through CELITE® (bag filter) into a clean drum. The reactor was rinsed with isopropyl acetate (7 kg) and the rinse is transferred to the drum. The organic phase was then concentrated under vacuum (25-28 inches of Hg) while maintaining reactor jacket temperature at 45-50° C. to afford 26 as an oil (˜10 L). Additional DIPEA (280 mL) was added and the vacuum distillation was continued (jacket temperature 50-55° C.) until no more distillate was collected. (batch volume ca. 7 L).
step 3—To the concentrated oil from step 2 containing 26 was added TEA (2.34 kg) and TEA-trihydrofluoride (1.63 kg). The mixture was heated to 85° C. for 2 h. The batch was sampled to monitor the progress of the reaction by GC. After the reaction was complete conc. HCl (2.35 kg) was added to the mixture and the resulting mixture heated to ca. 90° C. (small amount of distillate collected). The reaction mixture was stirred at ca. 90° C. for 30 min and then saturated aqueous BaCl2solution (18.8 kg) was added. The resulting suspension was stirred at about 90° C. for 4 h. The resulting mixture was then azeotropically dried under a vacuum (9-10 inches of Hg) by adding slowly n-propanol (119 kg) while distilling off the azeotropic mixture (internal batch temperature ca. 85-90° C.). To the residual suspension was added toluene (33 kg) and vacuum distillation was continued to distill off residual n-propanol (and traces of water) to a minimum volume to afford 28.
step 4—To the residue from step 3 containing 28 was added MeCN (35 kg) and ca. 15 L was distilled out under atmospheric pressure. The reaction mixture was cooled to ca. 10° C. and then benzoyl chloride (8.27 kg) and DMAP (0.14 kg) are added. TEA (5.84 kg) was added slowly to the reaction mixture while cooling to maintain temperature below 40° C. The batch was aged at ca. 20° C. and the progress of the benzoylation is monitored by HPLC. After completion of the reaction, EtOAc (30 kg) was added to the mixture and the resulting suspension is stirred for about 30 min. The reaction mixture was filtered through a CELITE® pad (using a nutsche filter) to remove inorganic salts. The solid cake was washed with EtOAc (38 kg). The combined filtrate and washes were washed successively with water (38 kg), saturated NaHCO3 solution (40 kg) and saturated brine (44 kg). The organic phase was polish-filtered (through a cartridge filter) and concentrated under modest vacuum to minimum volume. IPA (77 kg) was added to the concentrate and ca. 25 L of distillate was collected under modest vacuum allowing the internal batch temperature to reach ca. 75° C. at the end of the distillation. The remaining solution was then cooled to ca. 5° C. over 5 h and optionally aged overnight. The precipitate was filtered and washed with of cold (ca. 5° C.) IPA (24 kg). The product was dried under vacuum at 60-70° C. to afford 6.63 kg (70.7% theory of 10 which was 98.2% pure by HPLC.
EXAMPLE 1 Benzoic acid 3-benzoyloxy-5-(4-benzoylamino-2-oxo-2H-pyrimidin-1-yl)-4-fluoro-4-methyl-tetrahydro-furan-2-ylmethyl ester (14)

Trifluoroethanol (4.08 kg) is added slowly to a cold solution (−15° C.) of RED-AL® solution (12.53 kg) and toluene (21.3 kg) while maintaining the reaction temperature at or below −10° C. After warming up to RT (ca. 20° C.), the modified RED-AL reagent mixture (30.1 kg out of the 37.6 kg prepared) is added slowly to a pre-cooled solution (−15° C.) of fluorolactone dibenzoate 10 (10 kg) in DCM (94.7 kg) while maintaining reaction temperature at or below −10° C. After reduction of the lactone (monitored by in-process HPLC), a catalytic amount of tetrabutylammonium bromide (90 g) is added to the reaction mixture. Sulfiiryl chloride (11.86 kg) is then added while maintaining reaction temperature at or below 0° C. The reaction mixture is then heated to 40° C. until formation of the chloride is complete (ca. 4 h) or warmed to RT (20-25° C.) and stirred over night (ca. 16 h). The reaction mixture is cooled to about 0° C., and water (100 L) is added cautiously while maintaining reaction temperature at or below 15° C. The reaction mixture is then stirred at RT for ca. 1 h to ensure hydrolytic decomposition of excess sulfuryl chloride and the phases are separated. The organic layer is washed with a dilute solution of citric acid (prepared by dissolving 15.5 kg of citric acid in 85 L of water) and then with dilute KOH solution (prepared by dissolving 15 kg of 50% KOH in 100 L of water). The organic phase is then concentrated and solvents are replaced with chlorobenzene (2×150 kg) via atmospheric replacement distillation. The resulting solution containing 30 is dried azeotropically.
A suspension of N-benzoyl cytosine (8.85 kg), ammonium sulfate (0.07 kg) and hexamethyldisilazane (6.6 kg) in chlorobenzene (52.4 kg) is heated to reflux (ca. 135° C.) and stirred (ca. 1 h) until the mixture becomes a clear solution. The reaction mixture is then concentrated in vacuo to obtain 32 as a syrupy liquid. The anhydrous solution of 30 in chlorobenzene (as prepared) and stannic chloride (28.2 kg) is added to this concentrate. The reaction mixture is maintained at about 70° C. until the desired coupling reaction is complete (ca. 10 h) as determined by in-process HPLC. Upon completion, the reaction mixture is cooled to RT and diluted with DCM (121 kg). This solution is added to a suspension of solid NaHCO3 (47 kg) and CELITE® (9.4 kg) in DCM (100.6 kg). The resulting slurry is cooled to 10-15° C., and water (8.4 kg) is added slowly to quench the reaction mixture. The resulting suspension is very slowly (caution: gas evolution) heated to reflux (ca. 45° C.) and maintained for about 30 min. The slurry is then cooled to ca. 15° C. and filtered. The filter cake is repeatedly reslurried in DCM (4×100 L) and filtered. The combined filtrate is concentrated under atmospheric pressure (the distillate collected in the process is used for reslurrying the filter cake) until the batch temperature rises to about 90° C. and then allowed to cool slowly to about −5° C. The resulting slurry is aged for at least 2 h at −5° C. The precipitated product is filtered and washed with IPA (30 kg+20 kg), and oven-dried in vacuo at about 70° C. to afford 8.8 kg (57.3%) of 1-(2-deoxy-2-fluoro-2-methyl-3-5-O-dibenzoyl-β-D-ribofuranosyl)-N-4-benzoylcytosine (14, CAS Reg No. 817204-32-3) which was 99.3% pure.
EXAMPLE 2 4-Amino-1-(3-fluoro-4-hydroxy-5-hydroxymethyl-3-methyl-tetrahydro-furan-2-yl)-1H-pyrimidin-2-one (18)

A slurry of 14 (14.7 kg) in MeOH (92.6 kg) is treated with catalytic amounts of methanolic sodium methoxide (0.275 kg). The reaction mixture is heated to ca. 50° C. and aged (ca. 1 h) until the hydrolysis is complete. The reaction mixture is quenched by addition of isobutyric acid (0.115 kg). The resulting solution is concentrated under moderate vacuum and then residual solvents are replaced with IPA (80 kg). The batch is distilled to a volume of ca. 50 L. The resulting slurry is heated to ca. 80° C. and then cooled slowly to ca. 5° C. and aged (ca. 2 h). The precipitated product is isolated by filtration, washed with IPA (16.8 kg) and dried in an oven at 70° C. in vacuo to afford 6.26 kg (88.9%) of 18 which assayed at 99.43% pure.
.................................................................................
EXAMPLE 4 Preparation of 2′-deoxy-2′-fluoro-2′-C-methyluridine
2′-Deoxy-2′-fluoro-2′-C-methylcytidine (1.0 g, 1 eq) (Clark, J., et al., J. Med. Chem., 2005, 48, 5504-5508) was dissolved in 10 ml of anhydrous pyridine and concentrated to dryness in vacuo. The resulting syrup was dissolved in 20 ml of anhydrous pyridine under nitrogen and cooled to 0° C. with stirring. The brown solution was treated with benzoyl chloride (1.63 g, 3 eq) dropwise over 10 min. The ice bath was removed and stirring continued for 1.5 h whereby thin-layer chromatography (TLC) showed no remaining starting material. The mixture was quenched by addition of water (0.5 ml) and concentrated to dryness. The residue was dissolved in 50 mL of dichloromethane (DCM) and washed with saturated NaHCO3 aqueous solution and H2O. The organic phase was dried over NaSO4 and filtered, concentrated to dryness to give N4,3′,5′-tribenzoyl-2′-Deoxy-2′-fluoro-2′-C-methylcytidine (2.0 g, Yield: 91%).
N4,3′,5′-tribenzoyl-2′-Deoxy-2′-fluoro-2′-C-methylcytidine (2.0 g, 1 eq) was refluxed in 80% aqueous AcOH overnight. After cooling and standing at room temperature (15° C.), most of the product precipitated and then was filtered through a sintered funnel. White precipitate was washed with water and co-evaporated with toluene to give a white solid. The filtrate was concentrated and co-evaporated with toluene to give additional product which was washed with water to give a white solid. Combining the two batches of white solid gave 1.50 g of 3′,5′-dibenzoyl-2′-Deoxy-2′-fluoro-2′-C-methyluridine (Yield: 91%).
To a solution of 3′,5′-dibenzoyl-2′-Deoxy-2′-fluoro-2′-C-methyluridine (1.5 g, 1 eq) in MeOH (10 mL) was added a solution of saturated ammonia in MeOH (20 mL). The reaction mixture was stirred at 0° C. for 30 min, and then warmed to room temperature slowly. After the reaction mixture was stirred for another 18 hours, the reaction mixture was evaporated under reduced pressure to give the residue, which was purified by column chromatography to afford pure compound 2′-deoxy-2′-fluoro-2′-C-methyluridine (500 mg, Yield: 60%).
Example numbers 13-54 and 56-66 are prepared using similar procedures described for examples 5-8. The example number, compound identification, and NMR/MS details are shown below:
| entry 25 |
 |
| entry 251H NMR (DMSO-d6) δ 1.13-1.28 (m, 12H), 3.74-3.81 (m, 2H), 3.95-4.08 (m, 1H), 4.20-4.45 (m, 2H), 4.83-4.87 (m, 1H), 5.52-5.58 (m, 1H),5.84-6.15 (m, 3H), 7.17-7.23 (m, 3H), 7.35-7.39 (m, 2H), 7.54-7.57(m, 1H), 11.50 (s. 1H); MS, m/e 530.2 (M + 1)+ | ||||||
.........................................
Synthesis of diastereomerically pure nucleotide phosphoramidates.
Ross BS, Reddy PG, Zhang HR, Rachakonda S, Sofia MJ.
J Org Chem. 2011 Oct 21;76(20):8311-9. doi: 10.1021/jo201492m. Epub 2011 Sep 26.
J Org Chem. 2011 Oct 21;76(20):8311-9. doi: 10.1021/jo201492m. Epub 2011 Sep 26.
The HCV NS5B nucleoside and non-nucleoside inhibitors.
Membreno FE, Lawitz EJ.
Clin Liver Dis. 2011 Aug;15(3):611-26. doi: 10.1016/j.cld.2011.05.003. Review.
Clin Liver Dis. 2011 Aug;15(3):611-26. doi: 10.1016/j.cld.2011.05.003. Review.
Discovery of a β-d-2'-deoxy-2'-α-fluoro-2'-β-C-methyluridine nucleotide prodrug (PSI-7977) for the treatment of hepatitis C virus.
Sofia MJ, Bao D, Chang W, Du J, Nagarathnam D, Rachakonda S, Reddy PG, Ross BS, Wang P, Zhang HR, Bansal S, Espiritu C, Keilman M, Lam AM, Steuer HM, Niu C, Otto MJ, Furman PA.
J Med Chem. 2010 Oct 14;53(19):7202-18. doi: 10.1021/jm100863x.
J Med Chem. 2010 Oct 14;53(19):7202-18. doi: 10.1021/jm100863x.
Mechanism of activation of PSI-7851 and its diastereoisomer PSI-7977.
Murakami E, Tolstykh T, Bao H, Niu C, Steuer HM, Bao D, Chang W, Espiritu C, Bansal S, Lam AM, Otto MJ, Sofia MJ, Furman PA.
J Biol Chem. 2010 Nov 5;285(45):34337-47. doi: 10.1074/jbc.M110.161802. Epub 2010 Aug 26.
Michael J. Sofia,Donghui Bao, Wonsuk Chang, Jinfa Du, Dhanapalan Nagarathnam, Suguna Rachakonda, P. Ganapati Reddy, Bruce S. Ross, Peiyuan Wang, Hai-Ren Zhang, Shalini Bansal, Christine Espiritu, Meg Keilman, Angela M. Lam, Holly M. Micolochick Steuer, Congrong Niu, Michael J. Otto, and Phillip A. Furman; Discovery of a β-D-2’-Deoxy-2’-a-fluoro-2’-β-C-methyluridine Nucleotide Prodrug (PSI-7977) for the Treatment of Hepatitis C Virus; J. Med. Chem. 2010, 53, 7202–7218; Pharmasset, Inc.
Bruce S. Ross, P. Ganapati Reddy , Hai-Ren Zhang , Suguna Rachakonda , and Michael J. Sofia; Synthesis of Diastereomerically Pure Nucleotide Phosphoramidates; J. Org. Chem., 2011, 76 (20), pp 8311–8319; Pharmasset, Inc.
Peiyuan Wang, Byoung-Kwon Chun, Suguna Rachakonda, Jinfa Du, Noshena Khan, Junxing Shi, Wojciech Stec, Darryl Cleary, Bruce S. Ross and Michael J. Sofia; An Efficient and Diastereoselective Synthesis of PSI-6130: A Clinically Efficacious Inhibitor of HCV NS5B Polymerase; J. Org. Chem., 2009, 74 (17), pp 6819–6824;Pharmasset, Inc.
Jeremy L. Clark, Laurent Hollecker, J. Christian Mason, Lieven J. Stuyver, Phillip M. Tharnish, Stefania Lostia, Tamara R. McBrayer, Raymond F. Schinazi, Kyoichi A. Watanabe, Michael J. Otto, Phillip A. Furman, Wojciech J. Stec, Steven E. Patterson, and Krzysztof W. Pankiewicz; Design, Synthesis, and Antiviral Activity of 2‘-Deoxy-2‘-fluoro-2‘-C-methylcytidine, a Potent Inhibitor of Hepatitis C Virus Replication; J. Med. Chem., 2005, 48 (17), pp 5504–5508; Pharmasset, Inc
J Biol Chem. 2010 Nov 5;285(45):34337-47. doi: 10.1074/jbc.M110.161802. Epub 2010 Aug 26.
Michael J. Sofia,Donghui Bao, Wonsuk Chang, Jinfa Du, Dhanapalan Nagarathnam, Suguna Rachakonda, P. Ganapati Reddy, Bruce S. Ross, Peiyuan Wang, Hai-Ren Zhang, Shalini Bansal, Christine Espiritu, Meg Keilman, Angela M. Lam, Holly M. Micolochick Steuer, Congrong Niu, Michael J. Otto, and Phillip A. Furman; Discovery of a β-D-2’-Deoxy-2’-a-fluoro-2’-β-C-methyluridine Nucleotide Prodrug (PSI-7977) for the Treatment of Hepatitis C Virus; J. Med. Chem. 2010, 53, 7202–7218; Pharmasset, Inc.
Bruce S. Ross, P. Ganapati Reddy , Hai-Ren Zhang , Suguna Rachakonda , and Michael J. Sofia; Synthesis of Diastereomerically Pure Nucleotide Phosphoramidates; J. Org. Chem., 2011, 76 (20), pp 8311–8319; Pharmasset, Inc.
Peiyuan Wang, Byoung-Kwon Chun, Suguna Rachakonda, Jinfa Du, Noshena Khan, Junxing Shi, Wojciech Stec, Darryl Cleary, Bruce S. Ross and Michael J. Sofia; An Efficient and Diastereoselective Synthesis of PSI-6130: A Clinically Efficacious Inhibitor of HCV NS5B Polymerase; J. Org. Chem., 2009, 74 (17), pp 6819–6824;Pharmasset, Inc.
Jeremy L. Clark, Laurent Hollecker, J. Christian Mason, Lieven J. Stuyver, Phillip M. Tharnish, Stefania Lostia, Tamara R. McBrayer, Raymond F. Schinazi, Kyoichi A. Watanabe, Michael J. Otto, Phillip A. Furman, Wojciech J. Stec, Steven E. Patterson, and Krzysztof W. Pankiewicz; Design, Synthesis, and Antiviral Activity of 2‘-Deoxy-2‘-fluoro-2‘-C-methylcytidine, a Potent Inhibitor of Hepatitis C Virus Replication; J. Med. Chem., 2005, 48 (17), pp 5504–5508; Pharmasset, Inc
- Harrison C. Patent battle lines drawn as sofosbuvir gains approval. , Nat Rev Drug Discov , Volume 13 , Issue 1 , 2013 Dec 31
- Traynor K. Sofosbuvir approved for chronic hepatitis C infection. , Am J Health Syst Pharm , Volume 71 , Issue 2 , 2014 Jan 15
- Benhamou Y. HCV F1/F2 patients: treat now or continue to wait. , Liver Int , Volume 34 Suppl 1 , 2014 Feb
- Asselah T. HCV direct-acting antiviral agents: the best interferon-free combinations. , Liver Int , Volume 34 Suppl 1 , 2014 Feb
- Marcellin P. Second-wave IFN-based triple therapy for HCV genotype 1 infection: simeprevir, faldaprevir and sofosbuvir. , Liver Int , Volume 34 Suppl 1 , 2014 Feb
- Corouge M. Treatment of hepatitis C virus genotype 3-infection. , Liver Int , Volume 34 Suppl 1 , 2014 Feb
- Osinusi A. IFNL4-ΔG Genotype is Associated with Slower Viral Clearance in Hepatitis C, Genotype-1 Patients Treated with Sofosbuvir and Ribavirin. , J Infect Dis , 2013 Dec 23
- Alavian SM. Sofosbuvir has come out of the magic box. , Hepat Mon , Volume 13 , Issue 12 , 2013 Dec 16
- Ferguson MC. Sofosbuvir with ribavirin is safe and effective in hepatitis C genotype 1 with unfavourable pretreatment characteristics. , Evid Based Med , 2013 Dec 12
- Hunt S. Minimal Impact of Sofosbuvir and Ribavirin on Health Related Quality of Life in Chronic Hepatitis C (CH-C). , J Hepatol , 2013 Dec 10
SOVALDI is the brand name for sofosbuvir, a nucleotide analog inhibitor of HCV NS5B polymerase.
The IUPAC name for sofosbuvir is (S)-Isopropyl 2-((S)-(((2R,3R,4R,5R)-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-4-fluoro-3-hydroxy-4-methyltetrahydrofuran-2-yl)methoxy)-(phenoxy)phosphorylamino)propanoate. It has a molecular formula of C22H29FN3O9P and a molecular weight of 529.45. It has the following structural formula:
 |
Sofosbuvir is a white to off-white crystalline solid with a solubility of ≥ 2 mg/mL across the pH range of 2-7.7 at 37 oC and is slightly soluble in water.
SOVALDI tablets are for oral administration. Each tablet contains 400 mg of sofosbuvir. The tablets include the following inactive ingredients: colloidal silicon dioxide, croscarmellose sodium, magnesium stearate, mannitol, and microcrystalline cellulose. The tablets are film-coated with a coating material containing the following inactive ingredients: polyethylene glycol, polyvinyl alcohol, talc, titanium dioxide, and yellow iron oxide.
Elbasvir, MK 8742
1370468-36-2 cas
1370468-36-2 cas
methyl N-[(2S)-1-[(2S)-2-[4-[(6S)-3-[2-[(2S)-1-[(2S)-2-(methoxycarbonylamino)-3-methylbutanoyl]pyrrolidin-2-yl]-4H-imidazol-4-yl]-6-phenyl-6H-indolo[1,2-c][1,3]benzoxazin-10-yl]-2H-imidazol-2-yl]pyrrolidin-1-yl]-3-methyl-1-oxobutan-2-yl]carbamate
Methyl [(2S)-1-[(2S)-2-[4-[(6S)-3-[2-[(2S)-1-[(2S)-2-[(methoxycarbonyl)amino]-3-methylbutanoyl]pyrrolidin-2-yl]-1H-imidazol-4-yl]-6-phenylindolo[1,2-c][1,3]benzoxazin-10-yl]-1H-imidazol-2-yl]pyrrolidin-1-yl]-3-methyl-1-oxobutan-2-yl]carbamate
Methyl [(2S)-1-[(2S)-2-[4-[(6S)-3-[2-[(2S)-1-[(2S)-2-[(methoxycarbonyl)amino]-3-methylbutanoyl]pyrrolidin-2-yl]-1H-imidazol-4-yl]-6-phenylindolo[1,2-c][1,3]benzoxazin-10-yl]-1H-imidazol-2-yl]pyrrolidin-1-yl]-3-methyl-1-oxobutan-2-yl]carbamate
Carbamic acid, N,N‘-[[(6S)-6-phenyl-6H-indolo[1,2-c][1,3]benzoxazine-3,10-diyl]bis[1H-imidazole-5,2-diyl-(2S)-2,1-pyrrolidinediyl[(1S)-1-(1-methylethyl)-2-oxo-2,1-ethanediyl]]]bis-, C,C‘-dimethyl ester
Carbamic acid, N,N’-(((6S)-6-phenyl-6H-indolo(1,2-c)(1,3)benzoxazine-3,10-diyl)bis(1H-imidazole-5,2-diyl-(2S)-2,1-pyrrolidinediyl((1S)-1-(1-methylethyl)-2-oxo-2,1-ethanediyl)))bis-, C,C’-dimethyl ester
Dimethyl N,N’-(((6S)-6-phenylindolo(1,2-c)(1,3)benzoxazine-3,10-diyl)bis(1H-imidazole-5,2-diyl-(2S)-pyrrolidine-2,1-diyl((2S)-3-methyl-1-oxobutane-1,2-diyl)))dicarbamate
Methyl ((1S)-1-(((2S)-2-(4-((6S)-10-(2-((2S)-1-((2S)-2-((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1H-imidazol-4-yl)-6-phenyl-6H-indolo(1,2-c)(1,3)benzoxazin-3-yl)-1H-imidazol-2-yl)pyrrolidin-1-yl)carbonyl)-2-methylpropyl)carbamate
Dimethyl N,N’-(((6S)-6-phenylindolo(1,2-c)(1,3)benzoxazine-3,10-diyl)bis(1H-imidazole-5,2-diyl-(2S)-pyrrolidine-2,1-diyl((2S)-3-methyl-1-oxobutane-1,2-diyl)))dicarbamate
Methyl ((1S)-1-(((2S)-2-(4-((6S)-10-(2-((2S)-1-((2S)-2-((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-1H-imidazol-4-yl)-6-phenyl-6H-indolo(1,2-c)(1,3)benzoxazin-3-yl)-1H-imidazol-2-yl)pyrrolidin-1-yl)carbonyl)-2-methylpropyl)carbamate
MW 882.0171, C49 H55 N9 O7,
UNII-632L571YDK
MERCK-PHASE 2
HCV NS5A Inhibitors
MK-8742 is in phase II clinical development at Merck & Co. for the oral treatment of chronic hepatitis C infection in combination with MK-5172 and ribavirin. Phase I clinical trials are uongoing for the treatment of hepatitis C infected males. In 2013, breakthrough therapy designation was assigned to the compound.
MK-8742 is an inhibitor of Hepatitis C Virus (HCV) non-structural protein 5A (NS5A) that is being developed for the treatment of HCV infection. MK-8742 has broad, potent HCV genotypic activity in vitro against viral variants that are resistant to other NS5A inhibitors. MK-8742 exhibits potent antiviral activity during 5 days of monotherapy in patients with GT1 and GT3 chronic HCV infection. MK-8742 is currently in Phase IIB development.
ELBASVIR
MK-8742 is an inhibitor of Hepatitis C Virus (HCV) non-structural protein 5A (NS5A) that is being developed for the treatment of HCV infection. MK-8742 has broad, potent HCV genotypic activity in vitro against viral variants that are resistant to other NS5A inhibitors. MK-8742 exhibits potent antiviral activity during 5 days of monotherapy in patients with GT1 and GT3 chronic HCV infection. MK-8742 is currently in Phase IIB development.
………………
EXAMPLE 23
Preparation of Compound A

A mixture of Compound Int-19b (1.1 g, 3 mmol), (dibromomethyl)benzene (2.25 g, 9 mmol) and K2C03 (1.2 g, 9 mmol) in 15 mL of DMF was heated to 100 °C and allowed to stir at this temperature for 3 hours. The reaction mixture was cooled to room temperature, concentrated in vacuo and the residue obtained was dissolved with
dichloromethane and water. The aqueous phase was extracted with dichloromethane. The combined organic extracts were washed with brine, dried over Na2S04, filtered and concentrated in vacuo. The resulting residue was purified using flash column
chromatography on silica gel to provide Compound Int-23a (380 mg, 28 %) as a white solid. 1H MR (CDCI3): δ 7.72 (bs, 1 H), 7.44 – 7.46 (d, J= 8.4 Hz, 1 H), 7.21 – 7.28 (m, 3 H), 7.09 – 7.12 (m, 3 H), 7.04 (s, 1 H), 6.99 – 7.01 (bs, J= 6.8 Hz, 2 H), 6.78 (s, 1 H), 6.63 – 6.65 (d, J = 8.4 Hz, 1 H). MS (ESI)
m/e (M+H+): 456. Step B – Pre aration of Compound Int-23b

lnt-23a lnt-23b
To a solution of Int-23a (456 mg, 1.0 mmol) in 1,4-dioxane was added bis pinacol borate (2.2 mmol) , Pd(dppf)Cl2 (0.04 mmol) and KOAc (4 mmol). The reaction mixture was put under N¾ heated to 110°C and allowed to stir at this temperature for 3 hours. The reaction mixture was cooled to room temperature, concentrated in vacuo, and the residue obtained was purified using column chromatography on silica gel to provide Compound Int- 23b (590 mg, 87 % yield). 1H MR (CDC13): δ 8.13 (s, 1 H), 7.60 (d, J= 7.6 Hz, 1 H), 7.52 (d, J= 8.0 Hz, 1H), 7.36 – 7.39 (m, 1 H), 7.14 -7.19 (m, 4 H), 6.93 – 6.95 (m, 3 H), 6.90 (s, 1 H), 1.26 – 1.29 (s, 24 H). MS (ESI) m / e (M+H+): 550.
- Pre aration of Compound Int-23c
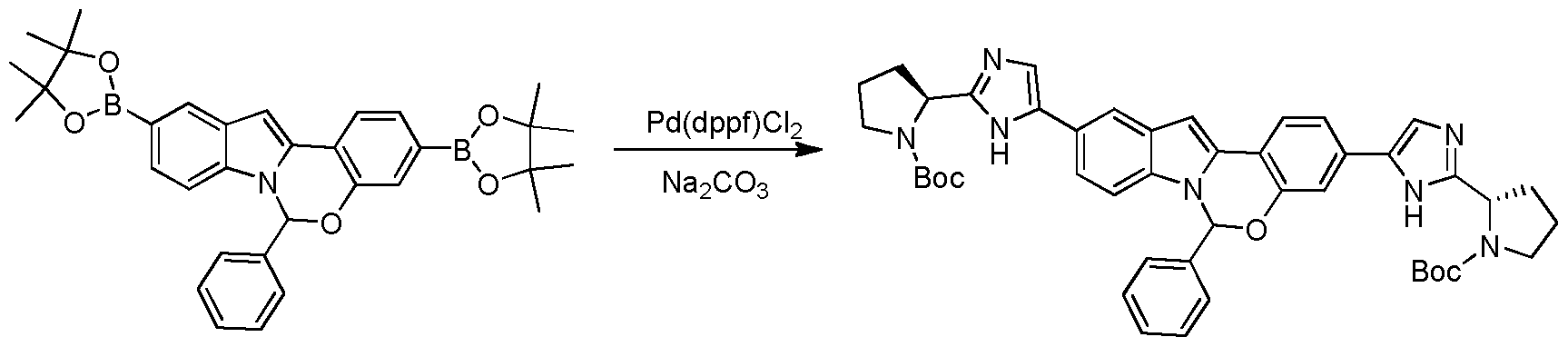
lnt-23b lnt-23c
A suspension of Int-23b (550 mg, 1.0 mmol), tert-butyl 2-(2-bromo-lH- imidazol-5-yl) pyrrolidine- 1-carboxylate (2.4 mmol), Pd(dppf) Cl2 (200 mg), Na2C03 (3 mmol) and in THF/H20 (10: 1, 33 mL) was allowed to stir at reflux for about 15 hours under N2. The reaction mixture was cooled to room temperature and filtered, and the filtrate was washed with water (50 mL) and extracted with EtOAc (100 mL). The organic extract was washed with brine, dried over anhydrous sodium sulfate, filtered and concentrated in vacuo. The resulting residue was purified using column chromatography on silica gel to provide Compound Int-23c (160 mg). MS (ESI) m / e (M+H+): 768.
Preparation of Compound Int-23d

Int-23c (0.10 g, 0.13 mmol) was added to HCl/CH3OH (5 mL, 3M) and the resulting reaction was allowed to stir at room temperature for about 3 hours. The reaction mixture was then concentrated in vacuo to provide Compound Int-23d, which was used without further purification. MS (ESI) m / e (M+H+): 568.
- Preparation of Compound A

To a solution of Int-23d (56.8 mg, 0.10 mmol), (S)-2- (methoxycarbonylamino)-3-methylbutanoic acid (35.0 mg, 0.20 mmol) and DIPEA (0.8 mmol) in CH3CN (1 mL) was added BOP (98 mg, 0.22 mmol). The resulting reaction was allowed to stir at room temperature and monitored using LCMS. After LCMS showed the starting material to be consumed, the reactionmixture was filtered, and the filtrate was purified using HPLC to provide Compound A as a white solid. 1H MR (MeOD): δ 7.94 (s,
1 H), 7.85 (d, J= 8.0 Hz, 1 H), 7.74 (s, 1 H), 7.63 (s, 1 H), 7.48 (s, 1 H), 7.35 – 7.37 (m, 2 H), 7.31 (s, 1 H), 7.17 – 7.18 (m, 4 H), 7.11 (s, 1 H), 6.96 – 6.98 (d, J = 7.6 Hz, 2 H), 5.09 – 5.17
(m, 2 H), 4.13 (t, J= 8.0 Hz, 2 H), 3.99 (bs, 2 H), 3.78 (bs, 2 H), 3.56 (s, 6 H), 2.44 – 2.47 (m,
2 H), 1.92 – 2.19 (m, 8 H), 0.77 – 0.85 (m, 12 H). MS (ESI) m / e (M+H+): 882.
The diastereomers were separated on a chiral SFC column: Isomer A: 1H NMR (MeOD): δ 8.08 (s, 1H), 7.91 – 7.93 (m, 1 H), 7.72 (s, 1 H), 7.56 (s, 1 H), 7.24 – 7.43 (m, 7 H), 7.19 (s, 1 H), 7.03 – 7.05 (m, 2 H), 5.16 – 5.24 (m, 2 H), 3.81 – 4.21 (m, 6 H), 3.62 (s, 6 H), 2.52 – 2.54 (m, 2 H), 2.00 – 2.25 (m, 8 H), 0.84 – 0.91 (m, 12 H). MS (ESI) m/z (M+H)+: 882.
Isomer B: 1H NMR (MeOD): δ 7.90 (s, 1 H), 7.81 – 7.83 (m, 1 H), 7.72 (s, 1 H), 7.62 (s, 1 H), 7.45 (s, 1 H), 7.14 – 7.33 (m, 6 H), 7.09 (s, 1 H), 6.93 – 6.95 (m, 2 H), 5.06 – 5.14 (m, 2 H), 3.71 – 4.11 (m, 6 H), 3.52 (s, 6 H), 2.41 – 2.44 (m, 2 H), 1.90 – 2.15 (m, 8 H), 0.74 – 0.86 (m, 12 H). MS (ESI) m/z (M+H)+: 882.
……………………..
Discovery of MK-8742: An HCV NS5A inhibitor with broad genotype activity
ChemMedChem 2013, 8(12): 1930
ChemMedChem 2013, 8(12): 1930
The NS5A protein plays a critical role in the replication of HCV and has been the focus of numerous research efforts over the past few years. NS5A inhibitors have shown impressive in vitro potency profiles in HCV replicon assays, making them attractive components for inclusion in all oral combination regimens. Early work in the NS5A arena led to the discovery of our first clinical candidate, MK-4882 [2-((S)-pyrrolidin-2-yl)-5-(2-(4-(5-((S)-pyrrolidin-2-yl)-1H-imidazol-2-yl)phenyl)benzofuran-5-yl)-1H-imidazole]. While preclinical proof-of-concept studies in HCV-infected chimpanzees harboring chronic genotype 1 infections resulted in significant decreases in viral load after both single- and multiple-dose treatments, viral breakthrough proved to be a concern, thus necessitating the development of compounds with increased potency against a number of genotypes and NS5A resistance mutations. Modification of the MK-4882 core scaffold by introduction of a cyclic constraint afforded a series of tetracyclic inhibitors, which showed improved virologic profiles. Herein we describe the research efforts that led to the discovery of MK-8742, a tetracyclic indole-based NS5A inhibitor, which is currently in phase 2b clinical trials as part of an all-oral, interferon-free regimen for the treatment of HCV infection.
see
Journal of Medicinal Chemistry (2014), 57(5), 1643-1672.
| WO2010111483A1 * | Mar 25, 2010 | Sep 30, 2010 | Merck Sharp & Dohme Corp. | Inhibitors of hepatitis c virus replication |
| US20070049593 * | Feb 23, 2005 | Mar 1, 2007 | Japan Tobacco Inc. | Tetracyclic fused heterocyclic compound and use thereof as HCV polymerase inhibitor |
GRAZOPREVIR
- Grazoprevir hydrate
- UNII-4O2AB118LA
- MK 5172
THERAPEUTIC CLAIM Antiviral
note........drug is k salt
MOLECULAR FORMULA C38H49N6O9SK
MOLECULAR WEIGHT804.99
MOLECULAR WEIGHT804.99
CHEMICAL NAMES
1. Cyclopropanecarboxamide, N-[[[(1R,2R)-2-[5-(3-hydroxy-6-methoxy-2-
quinoxalinyl)pentyl]cyclopropyl]oxy]carbonyl]-3-methyl-L-valyl-(4R)-4-hydroxy-L-prolyl-1-
amino-N-(cyclopropylsulfonyl)-2-ethenyl-, cyclic (1→2)-ether, hydrate (1 :1) (1R,2S)-
quinoxalinyl)pentyl]cyclopropyl]oxy]carbonyl]-3-methyl-L-valyl-(4R)-4-hydroxy-L-prolyl-1-
amino-N-(cyclopropylsulfonyl)-2-ethenyl-, cyclic (1→2)-ether, hydrate (1 :1) (1R,2S)-
2. (1aR,5S,8S,10R,22aR)-N-{(1R,2S)-1-[(cyclopropylsulfonyl)carbamoyl]-2-
ethenylcyclopropyl}-5-(1,1-dimethylethyl)-14-methoxy-3,6-dioxo-
1,1a,3,4,5,6,9,10,18,19,20,21,22,22a-tetradecahydro-8H-7,10-
methanocyclopropa[18,19][1,10,3,6]dioxadiazacyclononadecino[11,12-b]quinoxaline-8-
carboxamide hydrate
ethenylcyclopropyl}-5-(1,1-dimethylethyl)-14-methoxy-3,6-dioxo-
1,1a,3,4,5,6,9,10,18,19,20,21,22,22a-tetradecahydro-8H-7,10-
methanocyclopropa[18,19][1,10,3,6]dioxadiazacyclononadecino[11,12-b]quinoxaline-8-
carboxamide hydrate
MOLECULAR FORMULA C38H50N6O9S.H2O
MOLECULAR WEIGHT 784.92
MOLECULAR WEIGHT 784.92
SPONSOR Merck Sharp & Dohme Corp.
CAS REGISTRY NUMBER 1350462-55-3 HYDRATE, 1350514-68-9 (anhydrous)
WHO NUMBER
9857
9857
GRAZOPREVIR
MERCK
MK-5172 is in phase II clinical development at Merck & Co. for the oral treatment of chronic hepatitis C in combination with peginterferon and ribavirin and in combination with MK-8742. Phase I clinical trials are ongoing for the treatment of hepatitis C in patients with genotype 1 and genotype 3. In 2013, breakthrough therapy designation was assigned to the compound.
Discovery of MK-5172, a macrocyclic hepatitis C virus NS3/4a protease inhibitor
ACS Med Chem Lett 2012, 3(4): 332DOI: 10.1021/ml300017p
ACS Med Chem Lett 2012, 3(4): 332DOI: 10.1021/ml300017p
Development of a practical, asymmetric synthesis of the hepatitis c virus protease inhibitor MK-5172
Org Lett 2013, 15(16): 4174
Org Lett 2013, 15(16): 4174
References on MK-5172 hydrate:
[1]. Steven Harper , John A. McCauley , Michael T. Discovery of MK-5172, a Macrocyclic Hepatitis C Virus NS3/4a Protease Inhibitor. ACS Med. Chem. Lett., 2012, 3 (4), pp 332-336[2]. Summa V, Ludmerer SW, McCauley JA, MK-5172, a selective inhibitor of hepatitis C virus NS3/4a protease with broad activity across genotypes and resistant variants. Antimicrob Agents Chemother. 2012 Aug;56(8):4161-7.
[1]. Steven Harper , John A. McCauley , Michael T. Discovery of MK-5172, a Macrocyclic Hepatitis C Virus NS3/4a Protease Inhibitor. ACS Med. Chem. Lett., 2012, 3 (4), pp 332-336[2]. Summa V, Ludmerer SW, McCauley JA, MK-5172, a selective inhibitor of hepatitis C virus NS3/4a protease with broad activity across genotypes and resistant variants. Antimicrob Agents Chemother. 2012 Aug;56(8):4161-7.
WO2013142159
WO 2013106631
WO 2013101550
WO 2013028470
WO 2013028471
WO2013028465
WO 2010011566
Description:
IC50 Value: 7.4nM and 7nM for genotype1b and 1a respectively, in replicon system [1]
MK-5172 is a novel P2-P4 quinoxaline macrocyclic HCV NS3/4a protease inhibitor currently in clinical development.
in vitro: In biochemical assays, MK-5172 was effective against a panel of major genotypes and variants engineered with common resistant mutations observed in clinical studies with other NS3/4a protease inhibitors. In the replicon assay, MK-5172 demonstrated subnanomolar to low-nanomolar EC50s against genotypes 1a, 1b, and 2a [2].
in vivo: In rats, MK-5172 showed a plasma clearance of 28 ml/min/kg and plasma half-life of 1.4 hr. When dosed p.o. at 5 mg/kg, the plasma exposure of MK-5172 was good with an AUC of 0.7 uM.hr. The liver exposure of the compound was quite good (23 uM at 4 hr), and MK-5172 remained in liver 24 hr after a single p.o. 5 mg/kg dose. At 24 hr, the liver concentration of MK-5172 was 0.2 uM, which was over 25-fold higher than the IC50 in the replicon assay with 50% NHS. When dosed to dogs, MK-5172 showed low clearance of 5 ml/min/kg and a 3 hr half-life after i.v. 2 mg/kg dosing and had good plasma exposure (AUC=0.4 uM.hr) after a p.o. 1 mg/kg dose [1].
Clinical trial: Evaluation of Hepatic Pharmacokinetics for MK-5172 in Participants With Chronic Hepatitis C . Phase1
IC50 Value: 7.4nM and 7nM for genotype1b and 1a respectively, in replicon system [1]
MK-5172 is a novel P2-P4 quinoxaline macrocyclic HCV NS3/4a protease inhibitor currently in clinical development.
in vitro: In biochemical assays, MK-5172 was effective against a panel of major genotypes and variants engineered with common resistant mutations observed in clinical studies with other NS3/4a protease inhibitors. In the replicon assay, MK-5172 demonstrated subnanomolar to low-nanomolar EC50s against genotypes 1a, 1b, and 2a [2].
in vivo: In rats, MK-5172 showed a plasma clearance of 28 ml/min/kg and plasma half-life of 1.4 hr. When dosed p.o. at 5 mg/kg, the plasma exposure of MK-5172 was good with an AUC of 0.7 uM.hr. The liver exposure of the compound was quite good (23 uM at 4 hr), and MK-5172 remained in liver 24 hr after a single p.o. 5 mg/kg dose. At 24 hr, the liver concentration of MK-5172 was 0.2 uM, which was over 25-fold higher than the IC50 in the replicon assay with 50% NHS. When dosed to dogs, MK-5172 showed low clearance of 5 ml/min/kg and a 3 hr half-life after i.v. 2 mg/kg dosing and had good plasma exposure (AUC=0.4 uM.hr) after a p.o. 1 mg/kg dose [1].
Clinical trial: Evaluation of Hepatic Pharmacokinetics for MK-5172 in Participants With Chronic Hepatitis C . Phase1
Hepatitis C virus (HCV) infection is a major health problem that leads to chronic liver disease, such as cirrhosis and hepatocellular carcinoma, in a substantial number of infected individuals. Current treatments for HCV infection include immunotherapy with recombinant interferon-α alone or in combination with the nucleoside analog ribavirin.
Several virally-encoded enzymes are putative targets for therapeutic intervention, including a metalloprotease (NS2-3), a serine protease (NS3), a helicase (NS3), and an RNA-dependent RNA polymerase (NS5B). The NS3 protease is located in the N-terminal domain of the NS3 protein. NS4A provide a cofactor for NS3 activity.
Potential treatments for HCV infection have been discussed in the different references including Balsano, Mini Rev. Med. Chem. 8(4):307-318, 2008, Rönn et al., Current Topics in Medicinal Chemistry 8:533-562, 2008, Sheldon et al., Expert Opin. Investig. Drugs 16(8):1171-1181, 2007, and De Francesco et al., Antiviral Research 58:1-16, 2003
Different HCV inhibitors are described in different publications. Macrocyclic compounds useful as inhibitors the HCV protease inhibitors are described in WO 06/119061, WO 7/015785, WO 7/016441, WO 07/148,135, WO 08/051,475, WO 08/051,477, WO 08/051,514, WO 08/057,209. Additional HCV NS3 protease inhibitors are disclosed in International Patent Application Publications WO 98/22496, WO 98/46630, WO 99/07733, WO 99/07734, WO 99/38888, WO 99/50230, WO 99/64442, WO 00/09543, WO 00/59929, WO 02/48116, WO 02/48172, British Patent No. GB 2 337 262, and U.S. Pat. No. 6,323,180.
...........................
nmr
13C NMR (100 MHz, DMSO-d6) δ 172.32, 170.63, 169.04, 159.86, 156.95, 154.74, 148.10, 140.41, 133.55 (2 signals), 128.94, 118.21, 117.58, 105.89, 74.88, 59.75, 58.71, 55.68, 54.13, 54.01, 40.13, 34.49, 34.04, 33.76, 32.68, 30.71, 30.43, 28.55, 27.69, 27.28, 26.38, 21.98, 18.49, 10.67, 5.69, 5.46; MS (ES+) m/z 767 (M+H)+
(1aR,5S,8S,10R,22aR)-5-tert-butyl-N-((1R,2S)-1-{[(cyclopropylsulfonyl)amino]carbonyl}-2-vinylcyclopropyl)-14-methoxy-3,6-dioxo-1,1a,3,4,5,6,9,10,18,19,20,21,22,22a-tetradecahydro-8H-7,10-methanocyclopropa[18,19][1,10,3,6]dioxadiazacyclononadecino[11,12-b]quinoxaline-8-carboxamide
......................
NMR OF GRAZOPREVIR K SALT
Potassium {[(1R,2S)-1-({[(1aR,5S,8S,10R,22aR)-5-tert-butyl-14-methoxy-3,6-dioxo-
1,1a,3,4,5,6,9,10,18,19,20,21,22,22a-tetradecahydro-8H-7,10-
methanocyclopropa[18,19][1,10,3,6]dioxadiazacyclononadecino[11,12-b]quinoxalin-8-
yl]carbonyl}amino)-2-ethenylcyclopropyl]carbonyl}(cyclopropylsulfonyl)azanide (15 K-salt).
1,1a,3,4,5,6,9,10,18,19,20,21,22,22a-tetradecahydro-8H-7,10-
methanocyclopropa[18,19][1,10,3,6]dioxadiazacyclononadecino[11,12-b]quinoxalin-8-
yl]carbonyl}amino)-2-ethenylcyclopropyl]carbonyl}(cyclopropylsulfonyl)azanide (15 K-salt).
1H NMR (400 MHz, DMSO-d6) δ 7.91 (br s, 1 H), 7.75 (d, J =
8.3 Hz, 1 H), 7.15 (m, 1 H), 7.04 (m, 1 H), 5.97 (m, 1 H), 5.73 (br s, 1 H), 4.96 (m, 1 H), 4.79 (apparent q, J = 9.3 Hz, 1 H), 4.26 (dd, J = 9.7, 7.7 Hz, 1 H), 4.20 (d, J = 11.3 Hz, 1 H), 4.14 (d, J = 8.8 Hz, 1 H), 3.90 (dd, J = 11.1, 3.2 Hz, 1 H), 3.86 (s, 3 H), 3.62 (m, 1 H), 2.86-2.60 (m, 3 H), 2.38 (m, 1 H), 2.21 (m, 1 H), 1.80-1.48 (m, 6 H), 1.42 (m, 5 H), 1.14 (m, 1 H), 0.95 (m, 10 H), 0.81 (m, 2 H), 0.72-0.50 (m, 3 H), 0.41 (m, 1 H) ppm.http://pubs.acs.org/doi/suppl/10.1021/ml300017p/suppl_file/ml300017p_si_001.pdf
8.3 Hz, 1 H), 7.15 (m, 1 H), 7.04 (m, 1 H), 5.97 (m, 1 H), 5.73 (br s, 1 H), 4.96 (m, 1 H), 4.79 (apparent q, J = 9.3 Hz, 1 H), 4.26 (dd, J = 9.7, 7.7 Hz, 1 H), 4.20 (d, J = 11.3 Hz, 1 H), 4.14 (d, J = 8.8 Hz, 1 H), 3.90 (dd, J = 11.1, 3.2 Hz, 1 H), 3.86 (s, 3 H), 3.62 (m, 1 H), 2.86-2.60 (m, 3 H), 2.38 (m, 1 H), 2.21 (m, 1 H), 1.80-1.48 (m, 6 H), 1.42 (m, 5 H), 1.14 (m, 1 H), 0.95 (m, 10 H), 0.81 (m, 2 H), 0.72-0.50 (m, 3 H), 0.41 (m, 1 H) ppm.http://pubs.acs.org/doi/suppl/10.1021/ml300017p/suppl_file/ml300017p_si_001.pdf
...............................................................
GRAZOPREVIR
(1aR,5S,8S,10R,22aR)-5-tert-Butyl-N-((1R,2S)-1-{[(cyclopropylsulfonyl)amino] carbonyl}-2-
vinylcyclopropyl)-14-methoxy-3,6-dioxo-1,1a,3,4,5,6,9,10,18,19,20,21,22,22a-tetradecahydro-8H-
7,10-methanocyclopropa[18,19][1,10,3,6]dioxadiazacyclononadecino[11,12-b]quinoxaline-8-
carboxamide (MK-5172, 15).
vinylcyclopropyl)-14-methoxy-3,6-dioxo-1,1a,3,4,5,6,9,10,18,19,20,21,22,22a-tetradecahydro-8H-
7,10-methanocyclopropa[18,19][1,10,3,6]dioxadiazacyclononadecino[11,12-b]quinoxaline-8-
carboxamide (MK-5172, 15).
1H NMR (400 MHz, CD3
OD) δ 7.79 (dd, J = 9.6, 1.8 Hz, 1 H), 7.23 (s, 1 H), 7.22 (m, 1 H), 7.10 (d, J = 9.6 Hz, 1 H), 6.01 (apparent t, J = 3.6 Hz, 1 H), 5.74 (m, 1 H), 5.24 (dd, J = 17.0 Hz, 1.6 Hz, 1 H), 5.11 (dd, J = 10.4 Hz, 1.6 Hz, 1 H), 4.49 (d, J = 11.2 Hz, 1 H), 4.40 (m, 2 H), 4.13 (dd, J = 12.0 Hz, 4.0 Hz, 1 H), 3.92 (s, 3 H), 3.76 (m, 1 H), 2.92 (m, 2 H), 2.85 (m, 1 H), 2.55 (dd, J = 13.6 Hz, 6.4 Hz, 1 H), 2.28 (m, 1 H), 2.18 (apparent q, J =8.8 Hz, 1 H), 1.85 (dd, J = 8.0 Hz, 5.6 Hz, 1 H), 1.73 (m, 2 H), 1.5 (m, 2 H), 1.40 (dd, J = 9.6 Hz, 5.6 Hz, 1 H), 1.3 (m, 2 H), 1.23 (m, 4 H), 1.08 (s, 9 H), 0.99 (m, 2 H), 0.89 (m, 3 H), 0.73 (m, 1 H), 0.49 (m, 1 H) ppm; HRMS (ESI) m/z 767.3411 [(M+H)+; calcd for C38H51N6O9S: 767.3433].http://pubs.acs.org/doi/suppl/10.1021/ml300017p/suppl_file/ml300017p_si_001.pdf
OD) δ 7.79 (dd, J = 9.6, 1.8 Hz, 1 H), 7.23 (s, 1 H), 7.22 (m, 1 H), 7.10 (d, J = 9.6 Hz, 1 H), 6.01 (apparent t, J = 3.6 Hz, 1 H), 5.74 (m, 1 H), 5.24 (dd, J = 17.0 Hz, 1.6 Hz, 1 H), 5.11 (dd, J = 10.4 Hz, 1.6 Hz, 1 H), 4.49 (d, J = 11.2 Hz, 1 H), 4.40 (m, 2 H), 4.13 (dd, J = 12.0 Hz, 4.0 Hz, 1 H), 3.92 (s, 3 H), 3.76 (m, 1 H), 2.92 (m, 2 H), 2.85 (m, 1 H), 2.55 (dd, J = 13.6 Hz, 6.4 Hz, 1 H), 2.28 (m, 1 H), 2.18 (apparent q, J =8.8 Hz, 1 H), 1.85 (dd, J = 8.0 Hz, 5.6 Hz, 1 H), 1.73 (m, 2 H), 1.5 (m, 2 H), 1.40 (dd, J = 9.6 Hz, 5.6 Hz, 1 H), 1.3 (m, 2 H), 1.23 (m, 4 H), 1.08 (s, 9 H), 0.99 (m, 2 H), 0.89 (m, 3 H), 0.73 (m, 1 H), 0.49 (m, 1 H) ppm; HRMS (ESI) m/z 767.3411 [(M+H)+; calcd for C38H51N6O9S: 767.3433].http://pubs.acs.org/doi/suppl/10.1021/ml300017p/suppl_file/ml300017p_si_001.pdf
................................
SYNTHESIS OF INTERMEDIATES Intermediates A
| Intermediate # | Structure | Name | Lit. Reference |
| A1 |  | (1R,2S)-1-Amino-N- (cyclopropylsulfonyl)-2- vinylcyclopropanecarboxamide hydrochloride | Wang et al., U.S. Pat. No. 6,995,174 |
Intermediate B1 3-methyl-N-({[(1R,2R)-2-pent-4-en-1-ylcyclopropyl]oxy}carbonyl)-L-valine

Step 1: [(1E)-hepta-1,6-dien-1-yloxy](trimethyl)silane
A solution (0.5 M) of butenyl magnesium bromide in THF (1.4 eq) was treated at −78° C. with Cu(I) Br.SMe2 (0.05 eq) and HMPA (2.4 eq). The mixture was stirred for 10 min, then a solution (1 M) of acrolein (1 eq) and TMSCl (2 eq) in THF was added over 1 h such that the internal temperature remained below −68° C. The resulting mixture was stirred at −78° C. for 2 h, then treated with excess Et3N and diluted with hexane. After reaching room temperature, the mixture was treated with a small portion of H2O and filtered through CELITE. The filtrate was washed 10 times with H2O and then with brine. The organic layer was dried, and the volatiles were removed to give a residue that was distilled under reduced pressure (20 mbar). The fraction collected at 80-86° C. contained the title compound (58%) as a colorless liquid. 1H NMR (400 MHz, CDCl3) δ 6.19 (d, J=11.6 Hz, 1H), 5.85-5.75 (m, 1H), 5.02-4.92 (m, 3H), 2.08-2.02 (m, 2H), 1.94-1.88 (m, 2H), 1.46-1.38 (m, 2H), 0.18 (s, 9H).
Step 2: trans-2-pent-4-en-1-ylcyclopropanol
A solution (0.45 M) of the preceding compound in hexane was treated with a solution (15%) of Et2Zn (1.2 eq) in toluene, and the resulting solution was cooled in an ice bath. Diiodomethane (1.2 eq) was added dropwise, then the solution was stirred for 1 h before being warmed to 20° C. Pyridine (6 eq) was added, and the slurry was stirred for 15 min then poured onto petroleum ether. The mixture was filtered repeatedly through CELITE until a transparent solution was obtained. This mixture was concentrated at 100 mbar, and the solution that remained (that contained trimethyl{[(trans)-2-pent-4-en-1-ylcyclopropyl]oxy}silane, toluene and pyridine) was further diluted with THF. The mixture was cooled to 0° C. then treated dropwise with a solution (1 M) of TBAF (1.2 eq) in THF. After 10 min, the mixture was allowed to warm to 20° C., and after a further 1 h was poured into H2O. The aqueous phase was extracted with EtOAc, and the combined organic extracts were washed with brine then dried. Removal of the volatiles afforded a residue that was purified by flash chromatography (eluent 0-66% Et2O/petroleum ether) to furnish the title compound (71%) as a colorless liquid. 1H NMR (400 MHz, CDCl3) δ 5.85-5.75 (m, 1H), 5.00 (dd, J=17.1, 1.6 Hz, 1H), 4.94 (br d, J=10.4 Hz, 1H), 3.20 (apparent dt, J=6.4, 2.5 Hz, 1H), 2.10-2.04 (m, 2H), 1.52-1.44 (m, 2H), 1.29-1.19 (m, 1H), 1.15-1.07 (m, 1H), 0.95-0.87 (m, 1H), 0.71-0.66 (m, 1H), 0.31 (apparent q, J=6.0 Hz, 1H).
Step 3: methyl 3-methyl-N-(oxomethylene)-L-valinate

A solution (0.39 M) of methyl 3-methyl-L-valinate in a 2:1 mixture of saturated aqueous NaHCO3 and CH2Cl2 was cooled in an ice bath and stirred rapidly. The mixture was treated with triphosgene (0.45 eq) in one portion, and the resulting mixture was stirred for 0.5 h. The reaction was diluted with CH2Cl2, and the layers were separated. The aqueous phase was extracted with CH2Cl2, then the combined organics were washed with brine and dried. Removal of the solvent gave the title compound as clear oil that was kept for 12 h under vacuum (0.1 mbar) then used directly in the subsequent step. 1H NMR (400 MHz, CDCl3) δ 3.79 (s, 3H), 3.75 (s, 1H), 1.00 (s, 9H).
Step 4: methyl 3-methyl-N-({[(1R,2R)-2-pent-4-en-1-ylcyclopropyl]oxy}carbonyl)-L-valinate and methyl 3-methyl-N-({[(1S,2S)-2-pent-4-en-1-ylcyclopropyl]oxy}carbonyl)-L-valinate

A solution (0.45 M) of trans-2-pent-4-en-1-ylcyclopropanol in toluene was treated with methyl 3-methyl-N-(oxomethylene)-L-valinate (1.1 eq) and then DMAP (1 eq). The resulting mixture was heated under reflux for 12 h then cooled to 20° C. H2O and EtOAc were added, and the organic layer was separated and washed with 1N HCl, brine and dried. Removal of the volatiles afforded a residue that was purified twice by flash chromatography (eluent 0-30% Et2O/petroleum ether). The first fractions contained methyl 3-methyl-N-({[(1R,2R)-2-pent-4-en-1-ylcyclopropyl]oxy}carbonyl)-L-valinate (38%) as an oil. MS (ES+) m/z 298 (M+H)+
The later fractions contained methyl 3-methyl-N-({[(1S,2S)-2-pent-4-en-1-ylcyclopropyl]oxy}carbonyl)-L-valinate (28%) as an oil. MS (ES+) m/z 298 (M+H)+
Step 5: 3-methyl-N-({[(1R,2R)-2-pent-4-en-1-ylcyclopropyl]oxy}carbonyl)-L-valine

A solution (0.1 M) of methyl 3-methyl-N-({[(1R,2R)-2-pent-4-en-1-ylcyclopropyl]oxy}carbonyl)-L-valinate in 2:1 mixture of MeOH/H2O was treated with LiOH.H2O (4 eq) and then heated at 60° C. for 4 h. The mixture was cooled and concentrated to half volume, then diluted with EtOAc and acidified with aqueous HCl (1 N). The organic layer was separated and washed with brine then dried. Removal of the volatiles afforded the title compound (98%) as an oil. MS (ES+) m/z 284 (M+H)+
Intermediates C Intermediate C1 methyl (4R)-4-[(3-chloro-7-methoxyquinoxalin-2-yl)oxy]-L-prolinate hydrochloride

Step 1: 6-methoxyquinoxaline-2,3-diol

A suspension of 4-methoxybenzene-1,2-diamine dihydrochloride in diethyl oxalate (8 eq) was treated with Et3N (2 eq) and then heated at 150° C. for 2 h. The mixture was cooled and filtered, and then the collected solid was washed with H2O and EtOH. The residue was dried to give the title compound (69%). MS (ES+) m/z 193 (M+H)+
Step 2: 3-chloro-6-methoxyquinoxalin-2-ol

A solution (1.53 M) of 6-methoxyquinoxaline-2,3-diol in DMF was treated with SOCl2 (1 eq) and heated at 110° C. After 1.5 h, the reaction mixture was cooled and poured into aqueous HCl (1 N). The resulting precipitate was filtered and washed with H2O and Et2O. The dried solid contained predominantly the title compound as a mixture with 6-methoxyquinoxaline-2,3-diol and 2,3-dichloro-6-methoxyquinoxaline. This material was used directly in the subsequent step. MS (ES+) m/z 211 (M+H)+
Step 3: 1-tert-butyl 2-methyl (2S,4R)-4-[(3-chloro-7-methoxyquinoxalin-2-yl)oxy]pyrrolidine-1,2-dicarboxylate

A solution (0.35 M) of 3-chloro-6-methoxyquinoxalin-2-ol in NMP was treated with Cs2CO3 (1.5 eq) and 1-tert-butyl 2-methyl (2S,4S)-4-{[(4-bromophenyl)sulfonyl]oxy}pyrrolidine-1,2-dicarboxylate (1.1 eq). The resulting mixture was stirred at 50° C. for 18 h, then a further portion (0.1 eq) of 1-tert-butyl 2-methyl (25,45)-4-{[(4-bromophenyl)sulfonyl]oxy}pyrrolidine-1,2-dicarboxylate was added. After stirring for 2 h, the mixture was cooled and diluted with H2O and EtOAc. The organic phases were washed with aqueous HCl (1 N), saturated aqueous NaHCO3 and brine. The dried organic phase was concentrated to a residue that was purified by flash-chromatography (0-60% EtOAc/petroleum ether) to give the title compound (35% for two steps) as a solid. MS (ES+) m/z 438 (M+H)+
Step 4: methyl (4R)-4-[(3-chloro-7-methoxyquinoxalin-2-yl)oxy]-L-prolinate hydrochloride

A solution (0.62 M) of 1-tert-butyl 2-methyl (2S,4R)-4-[(3-chloro-7-methoxyquinoxalin-2-yl)oxy]pyrrolidine-1,2-dicarboxylate in CH2Cl2 was treated with a solution (4 M) of HCl in dioxane (5 eq). The mixture was stirred at 20° C. for 2 h, then treated with a solution (4 M) of HCl in dioxane (2 eq). After 5 h, the reaction was judged complete and the mixture was concentrated under reduced pressure. The residue was triturated with Et2O to give the title compound (95%) as a solid. MS (ES+) m/z 338 (M+H)+
Example 1 Potassium {[(1R,2S)-1-({[(1aR,5S,8S,10R,22aR)-5-tert-butyl-14-methoxy-3,6-dioxo-1,1a,3,4,5,6,9,10,18,19,20,21,22,22a-tetradecahydro-8H-7,10-methanocyclopropa[18,19][1,10,3,6]dioxadiazacyclononadecino[11,12-b]quinoxalin-8-yl]carbonyl}amino)-2-vinylcyclopropyl]carbonyl}(cyclopropylsulfonyl)azanide

Step 1: methyl 3-methyl-N-({[(1R,2R)-2-pent-4-en-1-ylcyclopropyl]oxy}carbonyl)-L-valyl-(4R)-4-[(3-chloro-7-methoxyquinoxalin-2-yl)oxy]-L-prolinate

A solution (0.2 M) of methyl (4R)-4-[(3-chloro-7-methoxyquinoxalin-2-yl)oxy]-L-prolinate hydrochloride in DMF was treated with 3-methyl-N-({[(1R,2R)-2-pent-4-en-1-ylcyclopropyl]oxy}carbonyl)-L-valine (1.1 eq), DIEA (5 eq) and HATU (1.2 eq). The resulting mixture was stirred at 20° C. for 5 h, then diluted with EtOAc. The organic layer was separated and washed with aqueous HCl (1 N), saturated aqueous NaHCO3 and brine. The dried organic phase was concentrated under reduced pressure to give a residue that was purified by flash chromatography (eluent 10-30% EtOAc/petroleum ether) to furnish the title compound (96%) as an oil. MS (ES+) m/z 604 (M+H)+
Step 2: methyl 3-methyl-N-({[(1R,2R)-2-pent-4-en-1-ylcyclopropyl]oxy}carbonyl)-L-valyl-(4R)-4-[(7-methoxy-3-vinylquinoxalin-2-yl)oxy]-L-prolinate

A solution (0.1 M) of methyl 3-methyl-N-({[(1R,2R)-2-pent-4-en-1-ylcyclopropyl]oxy}carbonyl)-L-valyl-(4R)-4-[3-chloro-7-methoxyquinoxalin-2-yl)oxy]-L-prolinate in EtOH was treated with potassium trifluoro(vinyl)borate (1.5 eq) and triethylamine (1.5 eq). The resulting mixture was degassed, then PdCl2(dppf)-CH2Cl2 adduct (0.1 eq) was added. The mixture was heated under reflux for 1 h, then cooled to room temperature and diluted with H2O and EtOAc. The organic phase was separated, washed with H2O and brine then dried. Removal of the volatiles afforded a residue that was purified by flash chromatography (20-30% EtOAc/petroleum ether) to give the title compound as a yellow foam that was used directly in the subsequent step. MS (ES+) m/z 595 (M+H)+
Step 3: methyl (1aR,5S,8S,10R,18E,22aR)-5-tert-butyl-14-methoxy-3,6-dioxo-1,1a,3,4,5,6,9,10,20,21,22,22a-dodecahydro-8H-7,10-methanocyclopropa[18,19][1,10,3,6]dioxadiazacyclononadecino[11,12-b]quinoxaline-8-carboxylate

A solution (0.02 M) of methyl 3-methyl-N-({[(1R,2R)-2-pent-4-en-1-ylcyclopropyl]oxy}carbonyl)-L-valyl-(4R)-4-[(7-methoxy-3-vinylquinoxalin-2-yl)oxy]-L-prolinate in DCE was heated to 80° C. then treated with Zhan 1 catalyst (0.15 eq). The resulting mixture was stirred at 80° C. for 1 h, then cooled to room temperature and concentrated under reduced pressure. The residue was purified by flash chromatography (20-50% EtOAc/petroleum ether) to give the title compound (25% for 2 steps) as a foam. MS (ES+) m/z 567 (M+H)+
Step 4: methyl (1aR,5S,8S,10R,22aR)-5-tert-butyl-14-methoxy-3,6-dioxo-1,1a,3,4,5,6,9,10,18,19,20,21,22,22a-tetradecahydro-8H-7,10-methanocyclopropa[18,19][1,10,3,6]dioxadiazacyclononadecino[11,12-b]quinoxaline-8-carboxylate

A solution (0.05 M) of methyl (1aR,5S,8S,10R,18E,22aR)-5-tert-butyl-14-methoxy-3,6-dioxo-1,1a,3,4,5,6,9,10,20,21,22,22a-dodecahydro-8H-7,10-methanocyclopropa[18,19][1,10,3,6]dioxadiazacyclononadecino[11,12-b]quinoxaline-8-carboxylate in MeOH/dioxane (1:1 ratio) was treated with Pd/C (8% in weight). The resulting mixture was stirred under atmosphere of hydrogen for 4 h. The catalyst was filtered off, and the filtrate was concentrated under reduced pressure to give the title compound (98%) as a solid. MS (ES+) m/z 569 (M+H)+
Step 5: (1aR,5S,8S,10R,22aR)-5-tert-butyl-14-methoxy-3,6-dioxo-1,1a,3,4,5,6,9,10,18,19,20,21,22,22a-tetradecahydro-8H-7,10-methanocyclopropa[18,19][1,10,3,6]dioxadiazacyclononadecino[11,12-b]quinoxaline-8-carboxylic acid

A solution (0.1 M) of methyl (1aR,5S,8S,10R,22aR)-5-tert-butyl-14-methoxy-3,6-dioxo-1,1a,3,4,5,6,9,10,18,19,20,21,22,22a-tetradecahydro-8H-7,10-methanocyclopropa[18,19][1,10,3,6]dioxadiazacyclononadecino[11,12-b]quinoxaline-8-carboxylate in a 1:1 mixture of H2O/THF was treated with LiOH.H2O (3 eq). The resulting mixture was stirred at 20° C. for 18 h, acidified with aqueous HCl (0.2 M) and diluted with EtOAc. The organic phase was separated, washed with aqueous HCl (0.2 M) and brine then dried. Removal of the volatiles afforded the title compound (98%) as a solid. MS (ES+) m/z 555 (M+H)+
Step 6: (1aR,5S,8S,10R,22aR)-5-tert-butyl-N-((1R,2S)-1-{[(cyclopropylsulfonyl)amino]carbonyl}-2-vinylcyclopropyl)-14-methoxy-3,6-dioxo-1,1a,3,4,5,6,9,10,18,19,20,21,22,22a-tetradecahydro-8H-7,10-methanocyclopropa[18,19][1,10,3,6]dioxadiazacyclononadecino[11,12-b]quinoxaline-8-carboxamide

A solution (0.1 M) of (1aR,5S,8S,10R,22aR)-5-tert-butyl-14-methoxy-3,6-dioxo-1,1a,3,4,5,6,9,10,18,19,20,21,22,22a-tetradecahydro-8H-7,10-methanocyclopropa[18,19][1,10,3,6]dioxadiazacyclononadecino[11,12-b]quinoxaline-8-carboxylic acid in CH2Cl2 was treated with (1R,2S)-1-{[(cyclopropylsulfonyl)amino]carbonyl}-2-vinylcyclopropanaminium chloride (1.3 eq), DIEA (3 eq), DMAP (1.5 eq) and TBTU (1.45 eq). The resulting mixture was stirred at 20° C. for 18 h and then diluted with EtOAc. The solution was washed with aqueous HCl (0.2 M), saturated aqueous NaHCO3 and brine. The organic phases were dried and concentrated to give a residue that was purified by flash-chromatography (eluent 2.5% MeOH/CH2Cl2) to give the title compound (89%) as a solid. 13C NMR (100 MHz, DMSO-d6) δ 172.32, 170.63, 169.04, 159.86, 156.95, 154.74, 148.10, 140.41, 133.55 (2 signals), 128.94, 118.21, 117.58, 105.89, 74.88, 59.75, 58.71, 55.68, 54.13, 54.01, 40.13, 34.49, 34.04, 33.76, 32.68, 30.71, 30.43, 28.55, 27.69, 27.28, 26.38, 21.98, 18.49, 10.67, 5.69, 5.46; MS (ES+) m/z 767 (M+H)+
GRAZOPREVIR POTASSIUM
Step 7: potassium {[(1R,2S)-1-({[(1aR,5S,8S,10R,22aR)-5-tert-butyl-14-methoxy-3,6-dioxo-1,1a,3,4,5,6,9,10,18,19,20,21,22,22a-tetradecahydro-8H-7,10-methanocyclopropa[18,19][1,10,3,6]dioxadiazacyclononadecino[11,12-b]quinoxalin-8-yl]carbonyl}amino)-2-vinylcyclopropyl]carbonyl}(cyclopropylsulfonyl)azanide

The preceding material was taken up in EtOH and the resulting solution (0.025 M) was cooled to 0° C. A solution (0.02 M) of tert-BuOK (1.5 eq) in EtOH was added leading to the formation of a precipitate. The mixture was stirred at 20° C. for 18 h, then the solid was collected by filtration. This material was washed with EtOH and dried to give the title compound (93%) as a white crystalline solid. MS (ES+) m/z 767 (M+H)+http://www.google.nl/patents/US8080654
Merck reported interim data from the Phase 2 C-WORTHY study in April 2014 at the International Liver Congress (ILC) in London that evaluated the efficacy and safety of its two-drug regimen based on NS3/4A protease inhibitor MK-5172 and NS5A replication complex inhibitor MK-8742, given with or without ribavirin, in GT1 HCV patients with cirrhosis. The once-daily single pill (without ribavirin) showed a 98% SVR12 (12-week sustained virologic response) in genotype-1, treatment-naive patients. Merck will start the phase III clinical trials (NCT02105688, NCT02105662, NCT02105467 andNCT02105701) for the combination in June 2014.
20 BMS-791325, Beclabuvir


























20 BMS-791325, Beclabuvir
BMS-791325, Beclabuvir
IN PHASE 2 for Hepatitis C (HCV)
An NS5B inhibitor.
BMS-791325 preferably is

CAS
958002-33-0
958002-36-3 (as hydrochloride)
958002-36-3 (as hydrochloride)
C36 H45 N5 O5 S, 659.838
Cycloprop(d)indolo(2,1-a)(2)benzazepine-9-carboxamide, 12-cyclohexyl-N-((dimethylamino)sulfonyl)-4b,5,5a,6-tetrahydro-3-methoxy-5a-((3-methyl-3,8-diazabicyclo(3.2.1)oct-8-yl)carbonyl)-, (4bS,5aR)-
(4bS,5aR)-12-Cyclohexyl-N-(dimethylsulfamoyl)-3-methoxy-5a-((3-methyl-3,8-diazabicyclo(3.2.1)oct-8-yl)carbonyl)-4b,5,5a,6-tetrahydrocyclopropa(d)indolo(2,1-a)(2)benzazepine-9-carboxamide
(4bS,5aR)-12-Cyclohexyl-N-(dimethylsulfamoyl)-3-methoxy-5a-((3-methyl-3,8-diazabicyclo(3.2.1)oct-8-yl)carbonyl)-4b,5,5a,6-tetrahydrocyclopropa(d)indolo(2,1-a)(2)benzazepine-9-carboxamide
(1aR,12bS)-8-Cyclohexyl-N-(dimethylsulfamoyl)-11-methoxy-1a-[(3-methyl-3,8-diazabicyclo[3.2.1]oct-8-yl)carbonyl]-1,1a,2,12b-tetrahydrocyclopropa[d]indolo[2,1-a][2]benzazepine-5-carboxamide
Cycloprop [d] indolo [2, 1 -a] [2]benzazepine-5-carboxamide, 8-cyclohexyl-N- [(dimethylamino)sulfonyl]-l,la,2,12b-tetrahydro-ll-methoxy-la-[(3-methyl-3,8- diazabicyclo[3.2.1]oct-8-yl)carbonyl]-, (laR,12bS)-
Bristol-Myers Squibb (Originator)
RNA-Directed RNA Polymerase (NS5B) Inhibitors
UNII-MYW1X5CO9S
BMS-791325 is in phase II clinical studies at Bristol-Myers Squibb for the treatment of chronic hepatitis C. In 2013, the company received breakthrough therapy designation in the U.S. for the treatment of chronic hepatitis C in combination with daclatasvir and asunaprevir.
| Patent | WO 2007136982 |
Scheme 1.

N-protected piperazines can also be coupled to the intermediate indolobenzazepine acids and the resultant piperazine carboxamides can be deprotected using methods known in the art and derivatized using a variety of synthetic protocols, some illustrative examples of which are shown below (See Scheme 2).
Scheme 2.

An intermediate useful for the synthesis of some compounds of the invention involves the preparation of the tert-butyl ester indolobenzazepine shown in Scheme 3. Scheme 3.
t-Butylation either:

This methodology involves base catalyzed hydrolysis of the indole methyl ester shown, followed by its reaction with either thionyl chloride and potassium tertiary butoxide, or alkylation with silver carbonate and tertiary butyl bromides. The resultant compound can be transformed using chemistry analogous to that outlined previously to provide the mixed ester indolobenzazepines shown above.
Scheme 4.

Some examples exist as stereoisomeric mixtures. The invention encompasses all stereoisomers of the compounds. Methods of fractionating stereoisomeric mixtures are well known in the art, and include but are not limited to; preparative chiral supercritical fluid chromatography (SFC) and chiral high performance liquid chromatography (HPLC). An example using this approach is shown in scheme 5. Scheme 5.

An additional method to achieve such separations involves the preparation of mixtures of diastereomers which can be separated using a variety of methods known in the art. One example of this approach is shown below (Scheme 6).
Scheme 6.

Diastereomers separated by reverse phase HPLC
Some diastereomeric amides can be separated using reverse phase HPLC. After hydroysis, the resultant optically active acids can be coupled with bridged piperazine derivatives (Scheme 6). For example, O-(lH-benzotriazol-l-yl)-N,N, N',N'-tetramethyluronium tetrafluoroborate and diisopropyl ethyl amine in DMSO can be used to give the alkyl bridged piperazine carboxamides. Other standard acid amine coupling methods can also be used to give optically active carboxamides.

Schemes 7-9 illustrate other methods of making intermediates and compounds.

Scheme 8.

Scheme 9.

Biological Methods
The compounds demonstrated activity against HCV NS5B as determined in the following HCV RdRp assays.
DESCRIPTION OF SPECIFIC EMBODIMENTS
Unless otherwise specified, analytical LCMS data on the following intermediates and examples were acquired using the following columns and conditions. Stop time: Gradient time + 1 minute; Starting cone: 0% B unless otherwise noted; Eluent A: 5% CH3CN / 95% H2O with 10 mM NH4OAc (for columns A, D and E); 10 % MeOH / 90 % H2O with 0.1% TFA (for columns B and C); Eluent B: 95% CH3CN / 5% H2O with 10 mM NH4OAc (for columns A, D and E); 90 % MeOH / 10 % H2O with 0.1% TFA (for columns B and C); Column A:
Phenomenex lOμ 4.6 x 50 mm C18; Column B: Phenomenex C18 lOμ 3.0 x 50 mm; Column C: Phenomenex 4.6 x 50 mm C18 lOμ; Column D: Phenomenex Lina C18 5μ 3.0 x 50 mm; Column E: Phenomenex 5μ 4.6 x 50 mm Cl 8.
Intermediate 1

lH-Indole-6-carboxylic acid, 2-bromo-3-cyclohexyl-, methyl ester. Freshly recrystallized pyridinium tribromide (recrystallization from hot AcOH (5 mL per 1 g), rinsed with cold AcOH and dried under high vacuum over KOH) was added in portions (over 10 min.) to a stirring solution of methyl 3-cyclohexyl-lH-indole-6- carboxylate (60 g, 233 mmol) (prepared using procedures describe in WO2004/065367) in CHC1/THF (1: 1, 1.25 L) at 2o C. The reaction solution was stirred at 0-5 °C for 2.5h, and washed with sat. aq. NaHSO3 (1 L), 1 N HCl (1 L) and brine (1 L). The organic layer was dried (MgSO4) and concentrated. The resulting red oil was diluted with Et2θ and concentrated. The resulting pink solid was dissolved into Et2θ (200 mL) treated with hexanes (300 mL) and partially concentrated. The solids were collected by filtration and rinsed with hexanes. The mother liquor was concentrated to dryness and the procedure repeated. The solids were combined to yield lH-indole-6-carboxylic acid, 2-bromo-3-cyclohexyl-, methyl ester (64 g, 190 mmol, 82%) as a fluffy pink solid, which was used without further purification. IHNMR (300 MHz, CDCl3) δ 8.47 (br s, IH), 8.03 (d, J = 1.4 Hz, IH), 7.74 (dd, J = 1.4, 8.8 Hz, IH), 7.69 (d, J = 8.8 Hz, IH), 3.92 (s, 3H), 2.82 (tt, J = 3.7, 11.7 Hz, IH), 1.98 - 1.72 (m, 7H), 1.50 - 1.27 (m, 3H). 13CNMR (75 MHz, CDC13) δ 168.2, 135.6, 130.2, 123.1, 120.8, 120.3, 118.7, 112.8, 110.7, 52.1, 37.0, 32.2(2), 27.0(2), 26.1. LCMS: m/e 334 (M-H)", ret time 3.34 min, column A, 4 minute gradient.
Intermediate 2

lH-Indole-6-carboxylic acid, 2-bromo-3-cyclohexyl-. A solution of methyl 2- bromo-S-cyclohexyl-lH-indole-ό-carboxylate (20 g, 60 mmol) and LiOH (3.8 g, 160 mmol) in MeOΗ/TΗF/Η2O ( 1 : 1 : 1 , 300 mL) was heated at 90 °C for 2h. The reaction mixture was cooled in an ice/H2O bath, neutralized with IM HCl (-160 mL) diluted with H2O (250 mL) and stirred for Ih at rt. The precipitates were collected by filtration rinse with H2O and dried to yield lH-indole-6-carboxylic acid, 2-bromo-3- cyclohexyl- (quant.) which was used without further purification.
An alternative procedure that can by used to provide lH-indole-6-carboxylic acid, 2-bromo-3-cyclohexyl- is described below: A solution of methyl 2-bromo-3-cyclohexyl-lH-indole-6-carboxylate (117 g, 349 mmol) and LiOKH2O (26.4 g, 629 mmol) in MeOH/THF/H2O (1: 1: 1, 1.8 L) was heated at reflux for 3h. The reaction mixture was cooled in an ice/H2O bath to ~2 °C, neutralized with IM HCl (-650 mL) (added at such a rate that temperature did not exceed 5 °C), diluted with H2O (1 L) and stirred while warming to ambient temperature. The precipitates were collected by filtration rinsed with H2O and dried to yield the mono THF solvate of lH-indole-6-carboxylic acid, 2-bromo-3- cyclohexyl- (135.5 g, 345 mmol, 99%) as a yellow solid, which was used without further purification. IHNMR (300 MHz, CDCl3) δ 11.01 (br s, IH), 8.77 (s, IH), 8.07 (d, J = 1.5 Hz, IH), 7.82 (dd, J = 1.5, 8.8 Hz, IH), 7.72 (d, J = 8.8 Hz, IH), 3.84 - 3.74 (m, 4H), 2.89 (m, IH), 1.98 - 1.72 (m, HH), 1.50 - 1.24 (m, 3H). 13CNMR (75 MHz, CDC13) δ 172.7, 135.5, 130.7, 122.3, 120.9(2), 118.8, 113.3, 111.1, 67.9(2), 37.0, 32.2(2), 27.0(2), 26.1, 25.5(2). LCMS: m/e 320 (M-H)", ret time 2.21 min, column A, 4 minute gradient.
Intermediate 3

lH-Indole-6-carboxamide, 2-bromo-3-cyclohexyl-N-
[(dimethylamino)sulfonyl]-. l,l'-Carbonyldiimidazole (1.17 g, 7.2 mmol) was added to a stirred solution of 2-bromo-3-cyclohexyl-lH-indole-6-carboxylic acid (2.03 g, 6.3 mmol) in THF (6 mL) at 22 °C. The evolution of CO2 was instantaneous and when it slowed the solution was heated at 50°C for 1 hr and then cooled to 220C. N,N-Dimethylsulfamide (0.94 g, 7.56 mmol) was added followed by the dropwise addition of a solution of DBU (1.34 g ,8.8 mmol) in THF (4 mL). Stirring was continued for 24 hr. The mixture was partitioned between ethyl acetate and dilute HCl. The ethyl acetate layer was washed with water followed by brine and dried over Na2SO4. The extract was concentrated to dryness to leave the title product as a pale yellow friable foam, (2.0 g, 74 %, >90 % purity , estimated from NMR). 1H NMR (300 MHz, DMSO-D6) δ ppm 1.28 - 1.49 (m, 3 H) 1.59 - 2.04 (m, 7 H) 2.74 - 2.82 (m, 1 H) 2.88 (s, 6 H) 7.57 (dd, J=8.42, 1.46 Hz, 1 H) 7.74 (d, J=8.78 Hz, 1 H) 7.91 (s, 1 H) 11.71 (s, 1 H) 12.08 (s, 1 H).
An alternative method for the preparation of lH-indole-6-carboxamide, 2- bromo-3-cyclohexyl-N-[(dimethylamino)sulfonyl]- is described below.
To a 1 L four necked round bottom flask equipped with a mechanical stirrer, a temperature controller, a N2 inlet , and a condenser, under N2, was added 2-bromo-3- cyclohexyl-lH-indole-6-carboxylic acid (102.0 g, 0.259 mol) and dry TΗF (300 mL). After stirring for 10 min, CDI (50.3 g, 0.31 mol) was added portion wise. The reaction mixture was then heated to 50 oC for 2 h. After cooling to 30 oC, N,N- dimethylaminosulfonamide (41.7 g, 0.336 mol) was added in one portion followed by addition of DBU (54.1 mL, 0.362 mol) drop wise over a period of 1 h. The reaction mixture was then stirred at rt for 20 h. The solvent was removed in vacuo and the residue was partitioned between EtOAc and 1 Ν HCl (1 : 1, 2 L). The organic layer was separated and the aqueous layer was extracted with EtOAc (500 mL). The combined organic layers were washed with brine (1.5 L) and dried over MgSO4. The solution was filtered and concentrated in vacuo to give the crude product (111.0 g). The crude product was suspended in EtOAc (400 mL) at 60 oC. To the suspension was added heptane (2 L) slowly. The resulting suspension was stirred and cooled to 0 oC. It was then filtered. The filter cake was rinsed with small amount of heptane and house vacuum air dried for 2 days. The product was collected as a white solid (92.0 g, 83%). 1H ΝMR (MeOD, 300 MHz) δ 7.89 (s, H), 7.77 (d, J= 8.4 Hz, IH), 7.55 (dd, J= 8.4 and 1.8 Hz, IH), 3.01 (s, 6H), 2.73-2.95 (m, IH), 1.81-2.05 (m, 8H), 1.39-1.50 (m, 2H); m/z 429 (M +H)+. Intermediate 4

lH-Indole-6-carboxamide, 3-cyclohexyl-N-[(dimethylamino)sulfonyl]-2-(2- formyl-4-methoxyphenyl)-. A mixture of the 2-Bromo-3-cyclohexyl- N- [(dimethylamino)sulfonyl]-lH-indole-6-carboxamide (4.28g, 0.01 mol), 4-methoxy- 2-formylphenyl boronic acid (2.1%, 0.015 mol), 2-dicyclohexylphosphino-2',6'- dimethoxy-biphenyl (41 mg, 0.0001 mol), palladium acetate (11.2 mg), and finely ground potassium carbonate (4.24g, 0.02 mol) in toluene (30 mL) was stirred under reflux and under nitrogen for 30 min, at which time LC/MS analysis showed the reaction to be complete. The reaction mixture was then diluted with ethyl acetate and water, and then acidified with an excess of dilute HCl. The ethyl acetate layer was then collected and washed with dilute HCl, water and brine. The organic solution was then dried (magnesium sulfate), filtered and concentrated to give a gum. The gum was diluted with hexanes (250 ml) and ethyl acetate (25 mL), and the mixture was stirred for 20 hr at 22° C during which time the product was transformed into a bright yellow granular solid (4.8 g) which was used directly without further purification.
An alternative procedure for the preparation of lH-indole-6-carboxamide, 3- cyclohexyl-N-[(dimethylamino)sulfonyl]-2-(2-formyl-4-methoxyphenyl)- is provided below:
To a slurried solution of 2-bromo-3-cyclohexyl-N-[(dimethylamino)sulfonyl]- indole-6-carboxamide (54.0 g, 126 mmol), 4-methoxy-2-formylphenylboronic acid (29.5 g, 164 mmol) and LiCl (13.3 g, 315 mmol) in EtOH/toluene (1 : 1, 1 L) was added a solution of Na2CO3 (40.1 g, 379 mmol) in water (380 mL). The reaction mixture was stirred 10 min. and then Pd(PPh3)4 (11.3 g, 10.0 mmol) was added. The reaction solution was flushed with nitrogen and heated at 70 °C (internal monitoring) overnight and then cooled to rt. The reaction was diluted with EtOAc (1 L) and EtOH (100 mL), washed carefully with IN aqueous HCl (1 L) and brine (500 mL), dried (MgSO4), filtered and concentrated. The residual solids were stirred with Et20 (600 mL) for Ih and collected by filtration to yield lH-indole-6-carboxamide, 3- cyclohexyl-N-[(dimethylamino)sulfonyl]-2-(2-formyl-4-methoxyphenyl)- (52.8g, 109 mmol, 87%) as a yellow powder which was used without further purification. IHNMR (300 MHz, d6-DMSO) δ 11.66 (s, IH), 8.17 (s, IH), 7.75 (d, J = 8.4 Hz, IH), 7.74 (d, J = 8.4 Hz, IH), 7.59 (dd, J = 1.4, 8.4 Hz, IH), 7.23 - 7.16 (m, 2H), 7.08 (dd, J = 2.6, 8.4 Hz, IH), 6.54 (d, J = 8.8 Hz, IH), 3.86 (s, 3H), 3.22 - 3.08 (m, IH), 2.91 (s, 6H), 2.00 - 1.74 (m, 7H), 1.60 - 1.38 (m, 3H). 13CNMR (75 MHz, CDC13) δ 165.7, 158.8, 147.2, 139.1, 134.3, 132.0, 123.4, 122.0, 119.2, 118.2, 114.8, 112.3, 110.4, 109.8, 79.6, 45.9, 37.2(2), 34.7, 32.0(2), 25.9(2), 24.9. LCMS: m/e 482 (M- H)", ret time 2.56 min, column A, 4 minute gradient.
Intermediate 5

6H-Isoindolo[2,l-a]indole-3-carboxamide, 11-cyclohexyl-N-
[(dimethylamino)sulfonyl]-6-ethoxy-8-methoxy-. To a 5 L four necked round bottom flask equipped with a temperature controller, a condenser, a N2 inlet and a mechanical stirrer, was charged toluene (900 mL), EtOH (900 mL), 2-bromo-3- cyclohexyl-N^NjN-dimethylsulfamoyiyiH-indole-ό-carboxamide (90 g, 0.21 mol), 2-formyl-4-methoxyphenylboronic acid (49.2 g, 0.273 mol) and LiCl (22.1 g, 0.525 mol). The resulting solution was bubbled with Ν2 for 15 mins. A solution of Na2CO3 (66.8 g, 0.63 mol) in Η2O (675 mL) was added and the reaction mixture was bubbled with N2 for another (10 mins). Pd(PPh3)4 (7.0 g, 6.3 mmol) was added and the reaction mixture was heated to 70 °C for 20 h. After cooling to 35 °C, a solution of 1 N HCl (1.5 L) was added slowly. The resulting mixture was transferred to a 6 L separatory funnel and extracted with EtOAc (2 X 1.5 L). The combined organic extracts were washed with brine (2 L), dried over MgSO4, filtered and concentrated in vacuo to give a yellow solid, which was triturated with 20% EtOAc in hexane (450 mL, 50 °C to 0 °C) to give 3-cyclohexyl-N-(N,N-dimethylsulfamoyl)-2-(2-formyl-4- methoxyphenyl)-lH-indole-6-carboxamide(65.9 g) as a yellow solid. HPLC purity, 98%.
The mother liquid from the trituration was concentrated in vacuo. The residue was refluxed with EtOH (50 mL) for 3 h. The solution was then cooled to 0 °C. The precipitates were filtered and washed with cooled TBME (5 °C) (20 mL). The filter cake was house vacuum air dried to give a further quantity of the title compound as a white solid (16.0 g). HPLC purity, 99%. 1H NMR (CDC13, 300 MHz) δ 8.75 (s, IH), 7.96 (s, IH), 7.73 (d, J= 8.4 Hz, IH), 7.67 (d, J= 8.4 Hz, IH), 7.45 (dd, J= 8.4 and 1.4 Hz, IH), 7.09 (d, J= 2.2 Hz, IH), 6.98 (dd, J= 8.4 and 2.2 Hz, IH), 6.50 (s, IH), 3.86 (s, 3H), 3.05 (s, 6H), 2.92-3.13 (m, 3H), 1.85-1.93 (m, 7 H), 1.40-1.42 (m, 3H), 1.05 (t, J= 7.1 Hz, 3H). m/z 512 (M + H)+.
Intermediate 6

lH-indole-6-carboxamide, 3-cyclohexyl-N-[(dimethylamino)sulfonyl]-2-(2- formyl-4-methoxyphenyl)-. 1 l-cyclohexyl-N-(N,N-dimethylsulfamoyl)-6-ethoxy-8- methoxy-6H-isoindolo[2,l-a]indole-3-carboxamide was dissolved in THF (75 mL). To the solution was added a solution of 2 N HCl (300 mL). The mixture was vigorously stirred under N2 at rt for 16 h. The resulting suspension was filtered and washed with cooled TBME (2 X 30 mL). the filer cake was vacuum air dried overnight to give the title compound as a yellow solid. HPLC purity, 99% 1H NMR (DMSO-d6, 300 MHz) δ 11.65 (s, IH), 8.16 (s, IH), 7.76 (d, J= 5.9 Hz, IH), 7.73 (d, J= 5.9 Hz, IH), 7.58 (dd, J= 8.5 and 1.5 Hz, IH), 7.17-7.20 (m, 2H), 7.08 (dd, J = 8.5 and 1.4 Hz, IH), 6.55 (d, J= 8.6 Hz, IH), 3.86 (s, 3H), 3.14-3.18 (m, IH), 2.91 (s, 6H), 1.75-1.99 (m, 7H), 1.48-1.60 (m, 3H); m/z 484 (M + H)+.
Intermediate 7

7H-Indolo[2, 1-a] ' [2] benzazepine-6-carboxylic acid, 13-cyclohexyl-10- [[[(dimethylamino)sulfonyl] amino] carbonyl]-3-methoxy-, methyl ester. A mixture of the 3-cyclohexyl-N-(N,N-dimethylsulfamoyl)-2-(2-formyl-4-methoxyphenyl)-lH- indole-6-carboxamide (4.8g, 0.01 mol), methyl 2-(dimethoxyphosphoryl)acrylate (9.7 g, 0.02 mol) and cesium carbonate (7.1g, 0.02 mol) in DMF (28mL) was stirred for 20 hr at an oil bath temperature of 55 ° C. The mixture was poured into ice-water and acidified with dilute HCl to precipitate the crude product. The solid was collected, dried and flash chromatographed on Siθ2 (11Og) using an ethyl acetate and methylene chloride (1: 10) solution containing 2% acetic acid. Homogeneous fractions were combined and evaporated to afford the title compound as a pale yellow solid (3.9g, 71 % yield). MS: 552 (M=H+).
An alternate procedure for the preparation of 7H-indolo[2,l- a] [2]benzazepine-6-carboxylic acid, 13-cyclohexyl-10- [[[(dimethylamino)sulfonyl]amino]carbonyl]-3-methoxy-, methyl ester is provided below. A solution of l l-cyclohexyl-N-[(dimethylamino)sulfonyl]-6-hydroxy-8- methoxy-6H-isoindolo[2,l-a]indole-3-carboxamide (cyclic hemiaminal) (63.0 g, 130 mmol), methyl 2-(dimethoxyphosphoryl)acrylate (60 g, 261 mmol), cesium carbonate (106 g, 326 mmol) in DMF (400 mL) was heated at 60 °C (bath temp) for 4.5h. Additional methyl 2-(dimethoxyphosphoryl)acrylate (15 g, 65 mmol) and cesium carbonate (21.2 g, 65 mmol) were added and the reaction was heated at 60 °C overnight then and cooled to rt. The stirring reaction mixture was diluted with H2O (1 L), slowly neutralized with IN aqueous HCl (800 mL), stirred 3h, and then the precipitates were collected by filtration. The solids were triturated with Et20 (800 mL) and dried to yield methyl 7H-indolo[2,l-a][2]benzazepine-6-carboxylic acid, 13- cyclohexyl-10-[[[(dimethylamino)sulfonyl]amino]carbonyl]-3-methoxy-, methyl ester (70.2 g, 127 mmol, 98%) as a yellow solid which was used without further purification. IHNMR (300 MHz, CDC13) δ 8.67 (s, IH), 8.09 (s, IH), 7.86 (d, J = 8.4 Hz, IH), 7.80 (s, IH), 7.50 (d, J = 8.4 Hz, IH), 7.42 (d, J = 8.8 Hz, IH), 7.08 (dd, J = 2.6, 8.8 Hz, IH), 6.98 (d, J = 2.6 Hz, IH), 5.75 - 5.51 (m, IH), 4.29 - 4.01 (m, IH), 3.89 (s, 3H), 3.82 (s, 3H), 3.05 (s, 6H), 2.87 - 2.73 (m, IH), 2.11 - 1.12 (m, 10H). LCMS: m/e 550 (M-H)-, ret time 3.21 min, column A, 4 minute gradient.
Example 1

Cycloprop[d]indolo[2,l-a] [2]benzazepine-5-carboxamide, 8-cyclohexyl-N- [(dimethylamino)sulfonyl]-l,la,2,12b-tetrahydro-ll-methoxy-la-[(3-methyl-3,8- diazabicyclo[3.2.1]oct-8-yl)carbonyl]-, (+/-)-. TBTU (43.7 mg, 0.136mmol) and DIPEA (0.095 mL, 0.544 mmol) were added to a solution of (+/-) cycloprop[d]indolo[2,l-a][2]benzazepine-la(2H)-carboxylic acid, 8-cyclohexyl-5- [[[(dimethylamino)sulfonyl]amino]carbonyl]-l,12b-dihydro-l 1-methoxy- (50 mg, 0.0906 mmol) in DMSO (2.0 mL). The reaction mixture was stirred at rt for 15 min. 3-Methyl-3,8-diaza-bicyclo[3.2. l]octane dihydrochloride {J & W PharmLab, LLC Morrisville, PA 19067-3620}. (27.1 mg, 0. 136 mmol) was then added and the reaction mixture was stirred at rt for 3 hr. It was then concentrated and the residue was purified by preparative reverse phase HPLC to give the final product as a yellow solid, (32 mg, 46% yield). MS m/z 660(MH+), Retention time: 2.445 min IH NMR (300 MHz, MeOD) δ ppm 0.20 (m, 0.23 H) 1.11 - 2.25 (m, 15.77 H) 2.58 (m, 0.23 H) 2.69 (m, 0.77 H) 2.75 - 3.11 (m, 10 H) 3.28 - 3.75 (m, 5 H) 3.91 (s, 2.31 H) 3.92 (s, 0.69 H) 4.15 - 4.37 (m, 1 H) 4.68 (m ,br, 1 H) 4.94 - 5.00 (m, 0.23 H) 5.16 (d, J=15.00 Hz, 0.77 H) 7.00 - 7.09 (m, 1 H) 7.18 (d, J=2.56 Hz, 0.23 H) 7.21 (d, J=2.56 Hz, 0.77 H) 7.33 (d, J=8.41 Hz, 0.77 H) 7.35 (d, J=8.42 Hz, 0.23 H) 7.57 (dd, J=8.42, 1.46 Hz, 0.77 H) 7.62 (dd, J=8.78, 1.46 Hz, 0.23 H) 7.91 (d, J=8.42 Hz, 0.77 H) 7.93 (d, J=8.42 Hz, 0.23 H) 8.00 (s, 0.77 H) 8.07 (s, 0.23 H).
Example 4

Cycloprop[d]indolo[2,l-a] [2]benzazepine-5-carboxamide, 8-cyclohexyl-N- [(dimethylamino)sulfonylj '- 1 , Ia, 2, 12b-tetrahydro-ll-methoxy-la-[(8-methyl-3, 8- diazabicyclo[3.2.1]oct-3-yl)carbonyl]-, (+/-)-. To a solution of (+/-) cycloprop[d]indolo[2,l-a][2]benzazepine-5-carboxamide, 8-cyclohexyl-la-(3,8- diazabicyclo[3.2.1]oct-3-ylcarbonyl)-N-[(dimethylamino)sulfonyl]-l,la,2,12b- tetrahydro-11-methoxy- (54 mg, 0.071 mmol) in methanol (3 mL), paraformaldehyde (6.4 mg, 0.213 mmol), ZnCl2 (29 mg, 0.213 mmol) and
Na(CN)BH3 (13.4 mg, 0.213 mmol) were added. The resultant mixture was heated at 60°C for 2hr, and then cooled to rt. The solid present was removed by filtration, and the filtrate was concentrated under vacuum and the residue purified by preparative reverse phase HPLC to give the title compound as a light yellow colored solid, (37 mg, 67% yield). MS ml 660(MH+), Retention time: 2.495 min. IH NMR (500 MHz, MeOD) δ ppm 0.21 (m, 0.3 H) 1.13 (m, 0.3 H) 1.18 - 2.22 (m, 15.4 H) 2.58 (m, 0.3 H) 2.68 (m, 0.7 H) 2.76 - 3.11 (m, 11 H) 3.32 - 3.37 (m, 1 H) 3.63 (d, J=15.56 Hz, 0.7 H) 3.82 - 4.32 (m, 7.3 H) 4.88 - 4.92 (m, 0.3 H) 5.08 (d, J=15.56 Hz, 0.7 H) 7.00 - 7.08 (m, 1 H) 7.18 (d, J=2.14 Hz, 0.3 H) 7.21 (d, J=2.14 Hz, 0.7 H) 7.32 (d, J=8.55 Hz, 0.7 H) 7.35 (d, J=8.55 Hz, 0.3H) 7.57 (d, J=7.93 Hz, 0.7 H) 7.62 (dd, J=8.39, 1.37 Hz, 0.3 H) 7.91 (d, J=8.55 Hz, 0.7 H) 7.93 - 7.99 (m, 1 H) 8.09 (s, 0.3 H).
Example 6

Cycloprop [d] indolo [2, 1 -a] [2]benzazepine-5-carboxamide, 8-cyclohexyl-N- [(dimethylamino)sulfonyl]-l,la,2,12b-tetrahydro-ll-methoxy-la-[(3-methyl-3,8- diazabicyclo[3.2.1]oct-8-yl)carbonyl]-, (laR,12bS)-. To a solution of (-) cycloprop[d]indolo[2,l-a][2]benzazepine-la(2H)-carboxylic acid, 8-cyclohexyl-5- [[[(dimethylamino)sulfonyl]amino]carbonyl]-l,12b-dihydro-l 1-methoxy- (204 mg, 0.37 mmol) in DMSO (8.0 mL), TBTU (178 mg, 0.555 mmol) and DIPEA (0.39 mL, 2.22 mmol) were added. The reaction mixture was stirred at rt for 15 min. Then 3- methyl-3,8-diaza-bicyclo[3.2.1]octane dihydrochloride (111 mg, 0. 555 mmol) was added and the reaction mixture was stirred at rt for 2 hr. It was then concentrated and the residue was purified by preparative reverse phase HPLC to give a yellow solid as final TFA salt. (265 mg, 92% yield). Average Specific Rotation: -53.56° Solvent, MeOH.; Wavelength 589 nm; 50 cm cell. MS m/z 660(MH+), Retention time: 3.035 min. 1H NMR (300 MHz, MeOD) δ ppm 0.20 (m, 0.23 H) 1.11 - 2.25 (m, 15.77 H) 2.58 (m, 0.23 H) 2.69 (m, 0.77 H) 2.75 - 3.11 (m, 10 H) 3.28 - 3.75 (m, 5 H) 3.91 (s, 2.31 H) 3.92 (s, 0.69 H) 4.15 - 4.37 (m, 1 H) 4.68 (m ,br, 1 H) 4.94 - 5.00 (m, 0.23 H) 5.16 (d, J=15.00 Hz, 0.77 H) 7.00 - 7.09 (m, 1 H) 7.18 (d, J=2.56 Hz, 0.23 H) 7.21 (d, J=2.56 Hz, 0.77 H) 7.33 (d, J=8.41 Hz, 0.77 H) 7.35 (d, J=8.42 Hz, 0.23 H) 7.57 (dd, J=8.42, 1.46 Hz, 0.77 H) 7.62 (dd, J=8.78, 1.46 Hz, 0.23 H) 7.91 (d, J=8.42 Hz, 0.77 H) 7.93 (d, J=8.42 Hz, 0.23 H) 8.00 (s, 0.77 H) 8.07 (s, 0.23 H). An alternate procedure for the synthesis of cycloprop[d]indolo[2,l- a][2]benzazepine-5-carboxamide, 8-cyclohexyl-N-[(dimethylamino)sulfonyl]- l,la,2,12b-tetrahydro-l l-methoxy-la-[(3-methyl-3,8-diazabicyclo[3.2.1]oct-8- yl)carbonyl]-, (laR,12bS)-rel-(-)-is provided below. To a mixture of (-) cycloprop[<i]indolo[2,l-α][2]benzazepine-la(2H)-carboxylic acid, 8-cyclohexyl-5- [[[(dimethylamino)sulfonyl]amino]carbonyl]-l,12b-dihydro-l 1-methoxy- (25.2 g, 45.68 mmol) and 3-methyl-3,8-diazabicyclo-[3.2.1]octane dihydrochloride (10.0 g, 50.22 mmol) in anhydrous MeCN (300 mL) was added DIPEA (23.62 g, 182.72 mmol) under N2. After 15 min, TBTU (16.12 g, 50.22 mmol) was added. The reaction solution was stirred for 30 min under N2. The ΗPLC indicated the disappearance of starting material. The solvent in the solution was evaporated to give a foam. This was dissolved in EtOAc (2.5 L), washed with H2O (1.5 L), H2O/brine (8:2) (1.5 L), brine (1.5 L), dried over Na2SO4 and evaporated to give 28.8 g of crude product. This solid was pooled with 45.4 g of material obtained from five separated reactions to afford a total of 74.2 g of crude product. This was passed through a pad of silica gel (E. Merck 230-400 mesh, 1 kg), eluting with MeOH/CH2Cl2 (2.5:97.5). After evaporation, it gave a foam, which was treated with EtOAc and hexane to turn into a solid. After drying at 50 °C under vacuum for 7 h, the GC analysis indicated it has 1.4% each of EtOAc and hexane. After further drying at 61-64 °C, the GC analysis indicated it still has 1.0% of hexane and 1.4% of EtOAc. The product was dissolved in Et2O and slowly evaporated in vacuum three times, dried at 60 °C under vacuum for 3 h to give 68.3 g. This was washed with H2O (900 mL) and redried at 68 °C under vacuum for 7 h to give 67.1 g (77% yield) of the compound of example 6. The GC analysis indicated it has 0.97% Of Et2O. HPLC conditions column: Cadenza CD-C18 3 x 250 mm; UV: 257 and 220 nm; 25 °C; flow rate: 0.5 mL/min; gradient time: 38 min, 0 - 80% B (0 - 35 min) and 80% B (35 - 38 min); solvent A: 25 nM CH3COONH4 at pH 4.7 in water, solvent B: MeCN. HPLC purity 99.7% (Rt 26.54 min); Chiral HPLC conditions column: Regis (S5S) Whelk-Ol 250 x 4.6 mm; UV 258nm; 35 °C; flow rate 2.0 mL/min; mobile phase C02/Me0H; gradient time 20 min, 30% MeOH (0 - 1 min), 30 - 48% MeOH (1 - 19 min), 48% MeOH (19 - 20 min). Chiral HPLC purity > 99.8% (Rt 16.60 min); LC/MS (ES+) 660.36 (M+H, 100); HRMS: calcd. 660.3220, found 660.3197; [α]D 25 C - 79.66 ° (c 1.06, MeOH); Anal. Calcd for C36H45N5O5S-O-O H2O»0.09 Et2O: C, 64.53; H, 7.00; N, 10.35; S, 4.74; H2O, 1.51; Et2O, 0.97. Found: C, 64.50; H, 7.12; N, 10.41; S, 5.14; H2O, 1.52; Et2O, 0.97. The absolute stereochemistry of cycloprop[d]indolo[2,l- a][2]benzazepine-5-carboxamide, 8-cyclohexyl-N-[(dimethylamino)sulfonyl]- l,la,2,12b-tetrahydro-l l-methoxy-la-[(3-methyl-3,8-diazabicyclo[3.2.1]oct-8- yl)carbonyl]-, (laR,12bS)-rel-(-)- is as drawn above, and was determined from an x- ray crystal structure obtained on the (R)-camphorsulfonic acid salt.
Additionally, the following salts were prepared: hydrochloride, phosphate, acetate, sulfate, camsylate, sodium, calcium, and magnesium. The hydrochloride salt had the following characteristics. DSC: small, broad endotherm from 25°C to 75°C, and potential melt/degradation endotherm with peak at temperatures ranging between 253 °C and 258 °C; TGA: Early weight loss from 25°C to 75°C ranging between 0.003% and 1.5%, and degradation weight loss starting at approximately 200°C.
| WO2006020082A1 * | Jul 15, 2005 | Feb 23, 2006 | Squibb Bristol Myers Co | Inhibitors of hcv replication |
| WO2006046030A2 * | Oct 25, 2005 | May 4, 2006 | Angeletti P Ist Richerche Bio | Tetracyclic indole derivatives as antiviral agents |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2008111978A1 | Mar 13, 2007 | Sep 18, 2008 | Squibb Bristol Myers Co | Cyclopropyl fused indolobenzazepine hcv ns5b inhibitors |
| WO2008112473A1 * | Mar 5, 2008 | Sep 18, 2008 | Squibb Bristol Myers Co | Compounds for the treatment of hepatitis c |
| WO2008112841A1 * | Mar 13, 2008 | Sep 18, 2008 | Squibb Bristol Myers Co | Compounds for the treatment of hepatitis c |
| WO2008112848A1 * | Mar 13, 2008 | Sep 18, 2008 | Squibb Bristol Myers Co | Compounds for the treatment of hepatitis c |
| WO2008112851A1 * | Mar 13, 2008 | Sep 18, 2008 | Squibb Bristol Myers Co | Cyclopropyl fused indolobenzazepine hcv inhibitors |
| WO2008112863A1 * | Mar 13, 2008 | Sep 18, 2008 | Squibb Bristol Myers Co | Compounds for the treatment of hepatitis c |
| WO2009067108A1 * | Nov 20, 2007 | May 28, 2009 | Squibb Bristol Myers Co | Cyclopropyl fused indolobenzazepine hcv ns5b inhibitors |
| WO2009067392A1 * | Nov 17, 2008 | May 28, 2009 | Squibb Bristol Myers Co | Cyclopropyl fused indolobenzazepine derivatives for the treatment of hepatitis c |
| WO2009067481A1 * | Nov 19, 2008 | May 28, 2009 | Squibb Bristol Myers Co | Compounds for the treatment of hepatitis c |
| WO2010080874A1 | Jan 7, 2010 | Jul 15, 2010 | Scynexis, Inc. | Cyclosporine derivative for use in the treatment of hcv and hiv infection |
| WO2013059265A1 * | Oct 17, 2012 | Apr 25, 2013 | Bristol-Myers Squibb Company | A compound for the treatment of hepatitis c |
| WO2014014885A1 * | Jul 16, 2013 | Jan 23, 2014 | Bristol-Myers Squibb Company | Novel methods and intermediates for the preparation of (4bs,5ar)-12-cyclohexyl-n-(n,n-dimethylsulfamoyl)-3-methoxy-5a-((1 r,5s) -3-methyl-3,8-diazabicyclo[3.2.1]octane-8-carbonyl)-4b,5,5a,6-tetrahydrobenzo [3,4]cyclopropa[5,6]azepino[1,2-a]indole-9-carboxamide |
| CN101679442B | Mar 13, 2008 | Feb 20, 2013 | 百时美施贵宝公司 | Compounds for the treatment of hepatitis c |
| EP2518073A1 * | Nov 19, 2008 | Oct 31, 2012 | Bristol-Myers Squibb Company | Compounds for the treatment of Hepatitis C |
LOPINAVIR
(2S)-N-[(2S,4S,5S)-5-[2-(2,6-dimethylphenoxy)acetamido]-4-hydroxy-1,6-diphenylhexan-2-yl]-3-methyl-2-(2-oxo-1,3-diazinan-1-yl)butanamide
[1S-[1R*,(R*),3R*,4R*]]-N-[4-[[(2,6-dimethyl-phenoxy)acetyl]amino]-3-hydroxy-5-phenyl-1-(phenylmethyl)pentyl]tetrahydro-alpha-(1-methylethyl)-2-oxo-1(2H)-pyrimidineacetamide
(2S,3S,5S)-2-(-2,6- dimethylphenoxyacetyl)-amino-3-hydroxy-5-(2-(1-tetrahydropyrimid-2-onyl)-3- methylbutanoyl)amino-1 ,6-diphenylhexane
628.8008
| Abbott Laboratories |
| CAS | 192725-17-0 |
|---|
| AHFS/Drugs.com | International Drug Names |
|---|---|
| MedlinePlus | a602015 |
| Pregnancy cat. | C (US) |
| Legal status | POM (UK) ℞-only (US) |
SYNONYMS
ABT-378, Aluviran, Koletra, ABT 378, 1mui, 2rkf, 2rkg, A 157378.0, RS-346
Molecular Formula: C37H48N4O5 Molecular Weight: 628.80082
...............
Inhibitors of human immunodeficiency virus (HIV) protease have been approved for use in the treatment of HIV infection for several years. A particularly effective and recently approved HIV protease inhibitor is (2S,3S,5S)-2-(-2,6- dimethylphenoxyacetyl)-amino-3-hydroxy-5-(2-(1-tetrahydropyrimid-2-onyl)-3- methylbutanoyl)amino-1 ,6-diphenylhexane (also known as lopinavir).

Lopinavir
Lopinavir is known to have utility for the inhibition of HIV protease and the inhibition of HIV infection. Lopinavir is particularly effective for the inhibition of HIV protease and for the inhibition of HIV infection when coadministered with ritonavir. Lopinavir, when combined with ritonavir, is also particularly effective for the inhibition of HIV infection when used in combination with one or more reverse transcriptase inhibitors and/or one or more other HIV protease inhibitors.
Lopinavir and processes for its preparation are disclosed in U.S. Patent No. 5,914,332, issued June 22, 1999, which is hereby incorporated herein by reference. This patent also discloses processes for preparing amorphous lopinavir.
Pharmaceutical compositions comprising lopinavir or a pharmaceutically acceptable salt thereof are disclosed in U.S. Patent No. 5,914,332, issued June 22, 1999; U.S. Patent Application No. 08/966,495, filed November 7, 1997; U.S. Provisional Application for Patent No. 60/177,020, filed January 19, 2000 and U.S. Patent Application No. 09/487,739, filed January 19, 2000, all of which are hereby incorporated herein by reference.
Lopinavir (ABT-378) is an antiretroviral of the protease inhibitor class. It is used against HIV infections as a fixed-dose combination with another protease inhibitor, ritonavir, under the trade names Kaletra (high-income countries) and Aluvia (low-income countries). It was first approved by the FDA on 15 September 2000.[1]
Lopinavir (ABT-378) is an antiretroviral of the protease inhibitor class. It is marketed by Abbott as Kaletra, a co-formulation with a sub-therapeutic dose of ritonavir, as a component of combination therapy to treat HIV/AIDS.
Retroviruses are those viruses which utilize a ribonucleic acid (RNA) intermediate and a RNA-dependent deoxyribonucleic acid (DNA) polymerase, reverse transcriptase, during their life cycle. Retroviruses include, but are not limited to, the RNA viruses of the Retroviridae family, and also the DNA viruses of the Hepadnavirus and Caulimovirus families. Retroviruses cause a variety of disease states in man, animals and plants. Some of the more important retroviruses from a pathological standpoint include human immunodeficiency viruses (HIV-1 and HIV-2), which cause acquired immune deficiency syndrome (AIDS) in man, human T-cell lymphotrophic viruses I, II, IV and V, which cause human acute cell leukemia, and bovine and feline leukemia viruses which cause leukemia in domestic animals.
Proteases are enzymes which cleave proteins at specific peptide bonds. Many biological functions are controlled or mediated by proteases and their complementary protease inhibitors. For example, the protease renin cleaves the peptide angiotensinogen to produce the peptide angiotensin I. Angiotensin I is further cleaved by the protease angiotensin converting enzyme (ACE) to form the hypotensive peptide angiotensin II. Inhibitors of renin and ACE are known to reduce high blood pressure in vivo. An inhibitor of a retroviral protease will provide a therapeutic agent for diseases caused by the retrovirus.
The genomes of retroviruses encode a protease that is responsible for the proteolytic processing of one or more polyprotein precursors such as the pol and gag gene products. See Wellink, Arch. Virol. 981 (1988). Retroviral proteases most commonly process the gag precursor into core proteins, and also process the pol precursor into reverse transciptase and retroviral protease. In addition, retroviral proteases are sequence specific. See Pearl, Nature 328 482 (1987).
The correct processing of the precursor polyproteins by the retroviral protease is necessary for the assembly of infectious virions. It has been shown that in vitro mutagenesis that produces protease-defective virus leads to the production of immature core forms which lack infectivity. See Crawford, J. Virol. 53 899 (1985); Katoh, et al., Virology 145 280 (1985). Therefore, retroviral protease inhibition provides an attractive target for antiviral therapy. See Mitsuya, Nature 325 775 (1987).
Current treatments for viral diseases usually involve administration of compounds that inhibit viral DNA synthesis. Current treatments for AIDS involve administration of compounds such as 3'-azido-3'-deoxythymidine (AZT), 2',3'-dideoxycytidine (DDC), 2',3'-dideoxyinosine (DDI), d4T and 3TC and compounds which treat the opportunistic infections caused by the immunosuppression resulting from HIV infection. None of the current AIDS treatments have proven to be totally effective in treating and/or reversing the disease. In addition, many of the compounds currently used to treat AIDS cause adverse side effects including low platelet count, renal toxicity and bone marrow cytopenia.
Recently the HIV protease inhibitors ritonavir, saquinavir and indinavir have been approved in the U.S. for treatment of HIV infections. However, there is a continuing need for improved HIV protease inhibitors.
Pharmacology
Lopinavir is highly bound to plasma proteins (98–99%).[2]
Reports are contradictory regarding lopinavir penetration into the cerebrospinal fluid (CSF). Anecdotal reports state that lopinavir cannot be detected in the CSF; however, a study of paired CSF-plasma samples from 26 patients receiving lopinavir/ritonavir found lopinavir CSF levels above the IC50 in 77% of samples.[3]
Clinical properties
Side effects, interactions, and contraindications have only been evaluated in the drug combination lopinavir/ritonavir.
Research
A 2014 study indicates that lopinavir is effective against the
human papilloma virus (HPV). The study used the equivalent of one tablet twice a day applied topically to the cervixes of women with high grade and low grade pre-cancerous conditions. After three months of treatment, 82.6% of the women who had high-grade disease had normal cervical conditions, confirmed by smears and biopsies.[4]
Lopinavir of Formula I is chemically [1S-[1R*,(R*),3R*,4R*]]-N-[4-[[(2,6-dimethyl-phenoxy)acetyl]amino]-3-hydroxy-5-phenyl-1-(phenylmethyl)pentyl]tetrahydro-alpha-(1-methylethyl)-2-oxo-1(2H)-pyrimidineacetamide and is indicated in combination with other antiretroviral agents for the treatment of HIV-infection.

U.S. Pat. No. 5,914,332 provides a process for preparing amorphous lopinavir which involves dissolving lopinavir in an organic solvent (for example, ethanol, isopropanol, acetone, or acetonitrile) and then adding the solution to water. For example, lopinavir is dissolved in ethanol (from about 2 to about 4 mL/g) and the ethanolic solution is added with stirring to water (from about 10 about 100 mL/g) to provide amorphous lopinavir. However, this process for the preparation of amorphous lopinavir is not effective on the kilogram scale and thus is not commercially suitable.
PCT Publication No. WO 01/074787 provides various crystalline Forms (Types I, II, III, IV) of solvated and non-solvated lopinavir. It further provides a process for the preparation of amorphous lopinavir which involves dehydration/desolvation of Type I hydrated crystal form/Type II solvated crystal forms.
PCT Publication Nos WO 2006/100552 and WO 2006/090264 provide process for the preparation of crystalline lopinavir.
Organic Process Research & Development, 3, 145-148 (1999), and Organic Process Research & Development, 4, 264-269 (2000); provide a crystallization process for the preparation of crystalline lopinavir which involves recrystallization from mixtures of ethyl acetate and heptane. However, the crystalline lopinavir obtained contains small amounts of solvents and removal of the final traces of solvents proved exceedingly difficult, and even extensive drying after milling (to reduce particle size) did not facilitate its complete removal. It further provides the crystallized product obtained contains appromixately 2% residual ethyl acetate which cannot be removed by further drying.
.....................................
https://www.google.com/patents/EP0882024A1?dq=5914332&ei=HkCVU9egNtOcugTls4HgDA
Scheme 1
3
Scheme I1A
\
Scheme MB
OH R2 O Scheme III
Scheme IV
10
......................................
http://www.google.com/patents/US20110224435
AMORPHOUS FORM
......................................................
http://www.google.com.ar/patents/WO2001074787A2?cl=en
POLYMORPHS
...................
http://www.google.com.ar/patents/US8445506
EXAMPLESExample 1
Thionyl chloride (18 ml) was added to the mixture of 2S-(1-tetrahydropyrimid-2-onyl)-3-methylbutanoic acid (25 gm), tetrahydrofuran (370 ml) and dimethylformamide (2 ml) at 0-10 deg C. and the mass was stirred for 1 hour 15 minutes. The mass was subjected to distillation under reduced pressure to remove excess thionyl chloride, n-heptane (45 ml) was added to the residue obtained and the solvent was distilled off. The reaction mass was slurried in dimethylformamide (105 ml). (2S,3S,5S)-2-(2,6-dimethylphenoxyacetyl)amino-3-hydroxy-5-amino-1,6-diphenylhexane (41 gm), imidazole (25 gm) and 4-(dimethylamino)pyridine (1.5 gm) were dissolved in ethyl acetate (420 ml). To the solution was added above slurried product at 0-10 deg C. The reaction mass was maintained for 14 hours and then ethyl acetate (165 ml) and water (250 ml) were added. The layers were separated, water (250 ml) was added to the organic layer and the pH was adjusted to 2.0-3.0 with dilute hydrochloric acid (6N HCl). The layers were separated, the organic layer was washed with aqueous sodium bicarbonate and then with water. The ethyl acetate was distilled off from the mass. The reaction mass was dissolved in ethyl acetate (80 ml) and n-heptane (80 ml) was added to the solution. The separated solid was stirred with ethyl acetate (290 ml) for 8 hours, filtered and dried the solid to obtain 33 gm of lopinavir ethyl acetate solvate
.................................
http://www.google.com/patents/US20130267547
........................................
Org. Proc. Res. Dev., 2000, 4 (4), pp 264–269
DOI: 10.1021/op990202j
http://pubs.acs.org/doi/abs/10.1021/op990202j
A large scale process for the synthesis of HIV protease inhibitor candidate ABT-378 has been developed which utilizes an intermediate common to the synthesis of ritonavir, Abbott's first generation compound. The synthesis relies on the sequential acylation of this intermediate which is carried through as a mixture of diastereomers until the penultimate step. A synthesis of acid 5, derived from l-valine, is also reported.
[1S-[1R*(R*),3R*,4R*]]-N-[4-[[(2,6-dimethylphenoxy)acetyl]amino]-3-hydroxy-5-phenyl-1-(phenylmethyl)pentyl]tetrahydro-α-(1-methylethyl)-2-oxo-1(2H)-pyrimidineacetamide (2).
A 500-mL, three-necked, round-bottomed flask equipped with mechanical stirring, ..........................DELETED.....................The solid product was washed with 30 mL of 1:1 EtOAc/heptane and dried in vacuo at 70 °C for 60 h, affording 18.8 g (89% yield) of ABT-378 2 as a colorless solid. Before crystallization crude 2 assayed as >93% pure by HPLC; after crystallization >99% purity was achieved.
mp (EtOAc), 124−127 °C. (uncorrected)
IR: 3413, 3335, 3289, 3060, 2966, 1671, 1650, 1624, 1545, 1520, 1453, 1189, 701 cm-1.
1H NMR (300 MHz): δ 7.30−7.13 (m, 10H), 7.02−6.92 (m, 3H), 6.86 (v br s, 1H), 5.68 (br s, 1H), 4.25 (m, 1H), 4.19 (app d, J = 10 Hz, 2H), 4.19 (m, 2H), 3.78 (m, app d sept, 1H), 3.12 (m, 1H), 3.06 (m, 2H), 2.97 (d, J = 7.6 Hz, 2H), 2.88 (m, 1H), 2.81 (app ABX dd, J = 14, 5.2 Hz, 1H), 2.68 (app ABX, dd, J = 14, 9.5 Hz, 1H), 2.23 (m, 1H), 2.18 (s, 6H), 1.83 (s, 1H), 1.74 (m, 2H), 1.53 (m, 1H), 1.28 (m, 2H), 0.83 (app t, J = 7 Hz, 6H).
13C NMR (75 MHz): δ 170.7, 168.8, 156.5, 154.2, 138.1, 138.0, 130.3, 129,3, 129.2, 129.0, 128.4, 128.2, 126.3, 126.0, 124.6, 70.2, 69.7, 63.1, 54.4, 48.7, 41.8, 41.1, 40.8, 40.0, 38.2, 25.4, 21.7, 19.6, 18.7, 16.1,
MS (ESI) 629 (M + H)+, 651 (M + Na)+.
Anal. Calcd for C37H48N4O5: C, 70.66; H, 7.69; N 8.91. Found: C, 70.26; H, 7.73; N 8.79.
[α]d20 = − 22.85 (c 0.4 MeOH).
- Crystallographic studies have shown, to our surprise, that 2 isolated by this crystallization method is not a solvate.
- The determination of the enantiomeric excess (% ee) for ABT-378 (2) can be done indirectly. Compound 17, which results from the acylation of 4 with the enantiomer of acid 5, is known to us, having been detected as an impurity in our process development.17 Compound 18 can only result from the acylation of the enantiomer of 4 (2R,3R,5R) with 5. The levels of 17/18 observed in 2 are typically <0.1%. Until there is a need for a more definitive assay, we assume this represents an upper limit to the amount of ent-2 present.
Enantiomeric excess is determined by HPLC (Chiracel OD column, elution with hexane: ethanol: trifluoroacetic acid (930: 70: 1). The desired l-isomer has a retention time of approximately 14 min; the d-isomer, 11.5 min.
References
- "FDA Approved Drug Products: Kaletra". Retrieved 30 April 2004.
- KALETRA (lopinavir/ritonavir) capsules; (lopinavir/ritonavir) oral solution. Prescribing information. April 2009
- Capparelli E, Holland D, Okamoto C, et al. (2005). "Lopinavir concentrations in cerebrospinal fluid exceed the 50% inhibitory concentration for HIV". AIDS (London, England) 19 (9).
- HIV drug used to reverse effects of virus that causes cervical cancer University of Manchester, 17 February 2014.
8-20-2003
|
Crystalline pharmaceutical
| |
12-27-2002
|
Compositions and methods for enhancing the bioavailability of pharmaceutical agents
| |
10-13-2000
|
PREGELATINIZED STARCH IN A CONTROLLED RELEASE FORMULATION
| |
6-20-1997
|
RETROVIRAL PROTEASE INHIBITING COMPOUNDS
|
8-8-2012
|
PROCESS FOR THE PREPARATION OF SUBSTANTIALLY PURE (2S,3S,5S)-5-AMINO-2-N,N-DIBENZYLAMINO-3-HYDROXY-1,6-DIPHENYLHEXANE
| |
11-12-2010
|
PRODRUGS OF HIV PROTEASE INHIBITORS
| |
5-19-2010
|
Prodrugs of HIV protease inhibitors
| |
5-7-2010
|
DIMETHYLPHENOXY MODULATORS OF VIRAL PROTEASE ACTIVITY AND/OR PARASITIC ENZYME ACTIVITY
| |
1-12-2007
|
Methods of treating cancer
| |
9-21-2005
|
Method to design therapeutically important compounds
| |
6-10-2005
|
Crystalline pharmaceutical
| |
3-9-2005
|
Crystalline pharmaceutical
| |
2-4-2005
|
Methods and compositions for the treatment or prevention of human immunodeficiency virus and related conditions using cyclooxygenase-2 selective inhibitors and antiviral agents
| |
8-27-2004
|
Methods of treating cancer
|
..........................
22 DASABUVIR, ABT 333
DASABUVIR, ABT 333
1132935-63-7
Non-nucleoside NS5B polymerase inhibitor
- Methanesulfonamide, N-(6-(5-(3,4-dihydro-2,4-dioxo-1(2H)-pyrimidinyl)-3-(1,1-dimethylethyl)-2-methoxyphenyl)-2-naphthalenyl)-
- N-(6-(3-tert-Butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl)methanesulfonamide
- Methanesulfonamide, N-(6-(5-(3,4-dihydro-2,4-dioxo-1(2H)-pyrimidinyl)-3-(1,1-dimethylethyl)-2-methoxyphenyl)-2-naphthalenyl)-
N-(6-(3-tert-Butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl)methanesulfonamide
- C26-H27-N3-O5-S
- 493.5813
- UNII-DE54EQW8T1,
Dasabuir (ABT-333), an oral non-nucleoside NS5B polymerase inhibitor, is a component of an all-oral hepatitis C treatment regimen under FDA review for the chronic Hepatitis C treatment.
On April 22, 2014, AbbVie submitted a New Drug Application (NDA) to the U.S. Food and Drug Administration (FDA) seeking approval for its investigational, all-oral, interferon-free regimen for the treatment of adult patients with chronic genotype 1 (GT1) hepatitis C virus (HCV) infection.
On April 22, 2014, AbbVie submitted a New Drug Application (NDA) to the U.S. Food and Drug Administration (FDA) seeking approval for its investigational, all-oral, interferon-free regimen for the treatment of adult patients with chronic genotype 1 (GT1) hepatitis C virus (HCV) infection.
................................
http://www.google.com/patents/WO2009039127A1?cl=en
Example 4A. Preparation of N-(6-(3-tert-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-l(2H)-yl)-2- methoxyphenyl)naphthalen-2-yl)methanesulfonamide (compound IB-LO-2.3).

[00768] Part A. Preparation of N-(6-bromonaphthalen-2-yl)methanesulfonamide. [00769] A solution of the product from Example 3, Part B (4.48g, 20.17mmol) in pyridine (10OmL) was treated drop wise with methanesulfonyl chloride (1.97mL, 2.89 g, 25.2mmol) followed by stirring at room temperature for Ih. Diluted with toluene and concentrated under vacuum twice. The residue was extracted with EtOAc and washed with water, IM citric acid and brine. Treated with Darco G-60, dried over Na2SO4, filtered through celite and concentrated under vacuum. Solid was triturated with ether- hexane, collected by filtration and dried under vacuum to give the title compound as a faint pink solid (3.32g, 55 %).
[00770] Part B. Preparation of N-(6-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)naphthalen-2-yl) methanesulfonamide .
[00771] A mixture of the product from Part A (1.0Og, 3.33mmol), bis(pincolato)diboron (1.27g,
5.00mmol), potassium acetate (0.98 g, 9.99mmol) and Combiphos Pd6 (84mg, 0.17mmol) in toluene
(22mL) was heated at reflux for 3h. Cooled and diluted with ethyl acetate and water. The mixture was treated with Darco G-60 and filtered through celite. The filtrate was washed with water and brine. Dried over Na2SO4, filtered and concentrated under vacuum. Oil was dissolved in ether and precipitated by addition of hexanes. The product was collected by filtration and washed with hexanes. Evaporation of the filtrate and purification by silica gel column chromatography eluting with EtOAc/hexanes. The title compound from crystallization and chromatography was obtained as a white solid (927mg, 80%).
[00772] Part C. Preparation of tert-butyl 3-tert-butyl-4-methoxy-5-(6-(methylsulfonamido) naphthalen-
2-yl)phenylcarbamate.
[00773] Combined the product from Example 3, Part H (87mg, 0.243mmol), the product from Part B
(169mg, 0.486mmol), toluene (1.0ml), ethanol (1.0ml) and sodium carbonate (0.243ml, 0.243mmol) in a sealed tube and de-gassed with N2 gas for 20min. Tetrakis(triphenylphosphine)palladium(0) (5.61mg,
4.86μmol) was added and de-gassing was continued another 5-10 min. Heated at 90-950C for 16h.
Cooled and concentrated under vacuum. Purification by silica gel column chromatography eluting with
EtOAc/hexanes gave the title compound (92.2mg, 76 %).
[00774] Part D. Preparation of N-(6-(3-tert-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-l(2H)-yl)-2- methoxyphenyl)naphthalen-2-yl)methanesulfonamide.
[00775] A solution of the product from Part C (90mg, 0.180mmol) in CH2Cl2 (2.0ml) was treated with trifluoroacetic acid (1.0ml, 12.98mmol) at room temperature for Ih. Concentrated under vacuum, dissolved residue in EtOAc, washed with 10% NaHCO3, and brine. Dried over Na2SO4, filtered and concentrated under vacuum. Dissolved in DMF (1.4ml) and cooled to -250C and added (E)-3-methoxy- acryloyl isocyanate (0.633ml, 0.361mmol) drop wise while maintaining the temperature below -1O0C. Warmed to room temperature and stirred for 2h. Poured into ether, washed with water, and brine. Dried over Na2SO4, filtered and concentrated under vacuum. Added a mixture OfH2SO4 (0.1ml, 1.876mmol), water (1.0ml) and EtOH (1.0ml) and stirred at 1000C 16h. Cooled and concentrated under vacuum. Poured into water, extracted with EtOAc, combined extracts and washed with brine. Dried over Na2SO4, filtered and concentrated under vacuum. Purification by silica gel column chromatography eluting with MeOH/CHCl3 gave the title compound (53mg, 59%). 1H NMR (300 MHz DMSO-J6) δ 1.42 (s, 9 H) 3.08 (s, 3 H) 3.25 (s, 3 H) 5.65 (d, J=7.72 Hz, 1 H) 7.34 (dd, J=15.81, 2.57 Hz, 2 H) 7.42 (dd, J=8.82, 1.84 Hz, 1 H) 7.65 - 7.76 (m, 2 H) 7.80 (d, J=8.09 Hz, 1 H) 7.96 (t, J= 8.27 Hz, 2 H) 8.02 (s, 1 H) 10.04 (s, 1 H) 11.41 (s, 1 H); MS (ESI+) m/z 494 (M+H)+; (ESI-) m/z 492 (M-H)".
[00776] Example 4B. Preparation of N-(6-(3-tert-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-l(2H)-yl)-2- methoxyphenyl)naphthalen-2-yl)methanesulfonamide (compound IB-LO-2.3).

[00777] Part A. Preparation of 2-tert-butyl-6-iodo-4-nitrophenol.
[00778] To the product from Example 3, Part E (4.5g, 23.05mmol) dissolved in MeOH (120ml) and water (3OmL) was added iodine monochloride (1.155ml, 23.05mmol) drop wise over a period of lOmin.
The mixture was stirred for 2h and diluted into IL of water and allowed to stand overnight. The solid material was collected by filtration and washed 3 x 5OmL with water and dried under vacuum overnight to give a tan solid (7.14g, 96%).
[00779] Part B. Preparation of l-tert-butyl-3-iodo-2-methoxy-5-nitrobenzene.
[0078O]To an ice bath cooled solution of the product from Part A (5.5g, 17.13mmol) in MTBE (15ml) in a 5OmL pressure vessel was added 2.0M trimethylsilyl diazomethane (12.85ml, 25.7mmol) followed by drop-wise addition of methanol (1.OmL) resulting in calm bubbling. The vessel was sealed and stirred at room temperature for 16h, cooled and the pressure was released. The solution was partitioned between
EtOAc and water. The organic layer was washed with 1.0M HCl, saturated potassium carbonate solution, and saturated NaCl. The organic layer was dried over sodium sulfate, filtered and concentrated to give a red oil that was used without purification (5.4g, 84%).
[00781] Part C. Preparation of 3-tert-butyl-5-iodo-4-methoxyaniline.
[00782] A mixture of the product from Part B (5.80g, 17.31mmol), ammonium chloride (1.389g,
26.0mmol), and iron (4.83g, 87mmol) in THF/MeOH/water (20OmL total, 2/2/1) was refluxed for 2h, cooled and filtered through Celite. The filtrate was evaporated and the residue was partitioned between water and EtOAc. The organic layer was washed with saturated brine, dried with sodium sulfate, filtered and evaporated to give a brown oil (5.28g, 100% yield).
[00783] Part D. Preparation of (E)-N-(3-tert-butyl-5-iodo-4-methoxyphenylcarbamoyl)-3-methoxy acrylamide.
[00784] To a solution of the product from Part C (3.05g, lOmmol) in DMF (50ml) at -20 0C under N2 was added at a fast drip a 0.4M solution in benzene of (E)-3-methoxyacryloyl isocyanate (50.0ml,
20.00mmol, prepared by the method of Santana et al., J. Heterocyclic. Chem. 36:293 (1999). The solution was stirred for 15min at -20 0C, warmed to room temperature for 45min and diluted with EtOAc. The organic was washed with water and brine. Dried over Na2SO4, filtered and concentrated to a brown solid. The residue was triturated in Et2θ/hexane to give a fine powder that was collected by filtration and dried under vacuum to give the title compound as a tan powder (2.46g, 57%).
[00785] Part E. Preparation of l-(3-tert-butyl-5-iodo-4-methoxyphenyl)dihydropyrimidine-2,4(lH,3H)- dione.
[00786] To a suspension of the product from Part D (2.46g, 5.69mmol) in ethanol (50ml) was added a solution of 5.5mL OfH2SO4 in 5OmL water and the mixture was heated at 1100C for 2.5h to give a clear solution. Cooled and diluted with 5OmL of water while stirring to give an off-white solid that was collected by filtration, washed with water and dried under vacuum to give the title compound (2.06g,
90%).
[00787]Part F. Preparation of N-(6-(3-tert-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-l(2H)-yl)-2- methoxyphenyl)naphthalen-2-yl)methanesulfonamide.
[00788] In a microwave tube, the product from Part E (104mg, 0.26mmol), the product from Example
4A, Part B (108mg, 0.31mmol), and 1.0M sodium carbonate solution (312μL, 0.31mmol) in 1: 1 ethanol- toluene (1.7mL) was degassed by nitrogen sparge for 15min. l,l'-Bis(diphenylphosphino) ferrocene palladium (II) chloride dichloromethane complex (9mg, 0.01 lmmol) was added, and degassing was continued for another 5min. The tube was sealed and heated in the microwave at 1000C for Ih. Diluted with dichloromethane and washed with IM citric acid solution and brine. The organic layer was then stirred with (3-mercaptopropyl) silica gel for Ih. Filtered through celite and concentrated under vacuum.
Triturated with ether, methanol, and then again with ether to give the title compound as a nearly white solid (32mg, 25 %). 1H NMR (300 MHz, DMSO-J6): δ 11.41 (d, J=I.84 Hz, 1 H) 10.04 (s, 1 H) 8.03 (s,
1 H) 7.96 (t, J=8.09 Hz, 2 H) 7.80 (d, J=8.09 Hz, 1 H) 7.63 - 7.79 (m, 2 H) 7.35 - 7.45 (m, 1 H) 7.37 (d,
J=2.57 Hz, 1 H) 7.32 (d, J=2.57 Hz, 1 H) 5.65 (dd, J=8.09, 2.21 Hz, 1 H) 3.25 (s, 3 H) 3.09 (s, 3 H) 1.43
(s, 9 H). MS (+ESI)m/z (rel abundance): 494 (100,M+H), 511 (90, M+NH4), 987 (20, 2M+H), 1009
(8, 2M+Na).
.........................
http://www.google.com/patents/WO2009039134A1?cl=en
Example 2A. Preparation of N-(6-(3-tert-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-l(2H)-yl)-2- methoxyphenyl)naphthalen-2-yl)methanesulfonamide (compound IB-LO-2.3).

[00511] Part A. Preparation of N-(6-bromonaphthalen-2-yl)methanesulfonamide. [00512] A solution of the product from Example 1, Part B (4.48g, 20.17mmol) in pyridine (10OmL) was treated drop wise with methanesulfonyl chloride (1.97mL, 2.89 g, 25.2mmol) followed by stirring at room temperature for Ih. Diluted with toluene and concentrated under vacuum twice. The residue was extracted with EtOAc and washed with water, IM citric acid and brine. Treated with Darco G-60, dried over Na2SO4, filtered through celite and concentrated under vacuum. Solid was triturated with ether- hexane, collected by filtration and dried under vacuum to give the title compound as a faint pink solid (3.32g, 55 %).
[00513] Part B. Preparation of N-(6-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)naphthalen-2-yl) methanesulfonamide .
[00514] A mixture of the product from Part A (1.0Og, 3.33mmol), bis(pincolato)diboron (1.27g,
5.00mmol), potassium acetate (0.98 g, 9.99mmol) and Combiphos Pd6 (84mg, 0.17mmol) in toluene
(22mL) was heated at reflux for 3h. Cooled and diluted with ethyl acetate and water. The mixture was treated with Darco G-60 and filtered through celite. The filtrate was washed with water and brine. Dried over Na2SO4, filtered and concentrated under vacuum. Oil was dissolved in ether and precipitated by addition of hexanes. The product was collected by filtration and washed with hexanes. Evaporation of the filtrate and purification by silica gel column chromatography eluting with EtOAc/hexanes. The title compound from crystallization and chromatography was obtained as a white solid (927mg, 80%).
[00515] Part C. Preparation of tert-butyl 3-tert-butyl-4-methoxy-5-(6-(methylsulfonamido) naphthalen-
2-yl)phenylcarbamate.
[00516] Combined the product from Example 1, Part H (87mg, 0.243mmol), the product from Part B
(169mg, 0.486mmol), toluene (1.0ml), ethanol (1.0ml) and sodium carbonate (0.243ml, 0.243mmol) in a sealed tube and de-gassed with N2 gas for 20min. Tetrakis(triphenylphosphine)palladium(0) (5.61mg,
4.86μmol) was added and de-gassing was continued another 5-10 min. Heated at 90-950C for 16h.
Cooled and concentrated under vacuum. Purification by silica gel column chromatography eluting with
EtOAc/hexanes gave the title compound (92.2mg, 76 %).
[00517]Part D. Preparation of N-(6-(3-tert-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-l(2H)-yl)-2- methoxyphenyl)naphthalen-2-yl)methanesulfonamide.
[00518] A solution of the product from Part C (90mg, 0.180mmol) in CH2Cl2 (2.0ml) was treated with trifluoroacetic acid (1.0ml, 12.98mmol) at room temperature for Ih. Concentrated under vacuum, dissolved residue in EtOAc, washed with 10% NaHCO3, and brine. Dried over Na2SO4, filtered and concentrated under vacuum. Dissolved in DMF (1.4ml) and cooled to -250C and added (E)-3-methoxy- acryloyl isocyanate (0.633ml, 0.361mmol) drop wise while maintaining the temperature below -1O0C. Warmed to room temperature and stirred for 2h. Poured into ether, washed with water, and brine. Dried over Na2SO4, filtered and concentrated under vacuum. Added a mixture Of H2SO4 (0.1ml, 1.876mmol), water (1.0ml) and EtOH (1.0ml) and stirred at 1000C 16h. Cooled and concentrated under vacuum. Poured into water, extracted with EtOAc, combined extracts and washed with brine. Dried over Na2SO4, filtered and concentrated under vacuum. Purification by silica gel column chromatography eluting with MeOH/CHCl3 gave the title compound (53mg, 59%). 1H NMR (300 MHz OMSO-d6) δ 1.42 (s, 9 H) 3.08 (s, 3 H) 3.25 (s, 3 H) 5.65 (d, J=7.72 Hz, 1 H) 7.34 (dd, J=15.81, 2.57 Hz, 2 H) 7.42 (dd, J=8.82, 1.84 Hz, 1 H) 7.65 - 7.76 (m, 2 H) 7.80 (d, J=8.09 Hz, 1 H) 7.96 (t, J= 8.27 Hz, 2 H) 8.02 (s, 1 H) 10.04 (s, 1 H) 11.41 (s, 1 H); MS (ESI+) m/z 494 (M+H)+; (ESI-) m/z 492 (M-H)".
[00519] Example 2B. Preparation of N-(6-(3-tert-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-l(2H)-yl)-2- methoxyphenyl)naphthalen-2-yl)methanesulfonamide (compound IB-LO-2.3).

[00520] Part A. Preparation of 2-tert-butyl-6-iodo-4-nitrophenol.
[00521] To the product from Example 1, Part E (4.5g, 23.05mmol) dissolved in MeOH (120ml) and water (3OmL) was added iodine monochloride (1.155ml, 23.05mmol) drop wise over a period of lOmin.
The mixture was stirred for 2h and diluted into IL of water and allowed to stand overnight. The solid material was collected by filtration and washed 3 x 5OmL with water and dried under vacuum overnight to give a tan solid (7.14g, 96%).
[00522]Part B. Preparation of l-tert-butyl-3-iodo-2-methoxy-5-nitrobenzene.
[00523] To an ice bath cooled solution of the product from Part A (5.5g, 17.13mmol) in MTBE (15ml) in a 5OmL pressure vessel was added 2.0M trimethylsilyl diazomethane (12.85ml, 25.7mmol) followed by drop-wise addition of methanol (1.OmL) resulting in calm bubbling. The vessel was sealed and stirred at room temperature for 16h, cooled and the pressure was released. The solution was partitioned between
EtOAc and water. The organic layer was washed with 1.0M HCl, saturated potassium carbonate solution, and saturated NaCl. The organic layer was dried over sodium sulfate, filtered and concentrated to give a red oil that was used without purification (5.4g, 84%).
[00524] Part C. Preparation of 3-tert-butyl-5-iodo-4-methoxyaniline.
[00525] A mixture of the product from Part B (5.8Og, 17.31mmol), ammonium chloride (1.389g,
26.0mmol), and iron (4.83g, 87mmol) in THF/MeOH/water (20OmL total, 2/2/1) was refluxed for 2h, cooled and filtered through Celite. The filtrate was evaporated and the residue was partitioned between water and EtOAc. The organic layer was washed with saturated brine, dried with sodium sulfate, filtered and evaporated to give a brown oil (5.28g, 100% yield).
[00526] Part D. Preparation of (E)-N-(3-tert-butyl-5-iodo-4-methoxyphenylcarbamoyl)-3-methoxy acrylamide.
[00527] To a solution of the product from Part C (3.05g, lOmmol) in DMF (50ml) at -20 0C under N2 was added at a fast drip a 0.4M solution in benzene of (E)-3-methoxyacryloyl isocyanate (50.0ml,
20.00mmol, prepared by the method of Santana et al., J. Heterocyclic. Chem. 36:293 (1999). The solution was stirred for 15min at -20 0C, warmed to room temperature for 45min and diluted with EtOAc. The organic was washed with water and brine. Dried over Na2SO4, filtered and concentrated to a brown solid. The residue was triturated in Et2O/hexane to give a fine powder that was collected by filtration and dried under vacuum to give the title compound as a tan powder (2.46g, 57%).
[00528] Part E. Preparation of l-(3-tert-butyl-5-iodo-4-methoxyphenyl)dihydropyrimidine-2,4(lH,3H)- dione.
[00529] To a suspension of the product from Part D (2.46g, 5.69mmol) in ethanol (50ml) was added a solution of 5.5mL OfH2SO4 in 5OmL water and the mixture was heated at 110°C for 2.5h to give a clear solution. Cooled and diluted with 5OmL of water while stirring to give an off-white solid that was collected by filtration, washed with water and dried under vacuum to give the title compound (2.06g,
[00530] Part F. Preparation of N-(6-(3-tert-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-l(2H)-yl)-2- methoxyphenyl)naphthalen-2-yl)methanesulfonamide.
[0053I]In a microwave tube, the product from Part E (104mg, 0.26mmol), the product from Example 2A, Part B (108mg, OJ lmmol), and 1.0M sodium carbonate solution (312μL, 0.31mmol) in 1:1 ethanol- toluene ( 1.7mL) was degassed by nitrogen sparge for 15min. 1 , 1 '-Bis(diphenylphosphino) ferrocene palladium (II) chloride dichloromethane complex (9mg, O.Ol lmmol) was added, and degassing was continued for another 5min. The tube was sealed and heated in the microwave at 1000C for Ih. Diluted with dichloromethane and washed with IM citric acid solution and brine. The organic layer was then stirred with (3-mercaptopropyl) silica gel for Ih. Filtered through celite and concentrated under vacuum. Triturated with ether, methanol, and then again with ether to give the title compound as a nearly white solid (32mg, 25 %). 1H NMR (300 MHz, OMSO-d6): δ 11.41 (d, J=1.84 Hz, 1 H) 10.04 (s, 1 H) 8.03 (s, 1 H) 7.96 (t, J=8.09 Hz, 2 H) 7.80 (d, J=8.09 Hz, 1 H) 7.63 - 7.79 (m, 2 H) 7.35 - 7.45 (m, 1 H) 7.37 (d, J=2.57 Hz, 1 H) 7.32 (d, J=2.57 Hz, 1 H) 5.65 (dd, J=8.09, 2.21 Hz, 1 H) 3.25 (s, 3 H) 3.09 (s, 3 H) 1.43 (s, 9 H). MS (+ESI) m/z (rel abundance): 494 (100, M+H), 511 (90, M+NH4), 987 (20, 2M+H), 1009 (8, 2M+Na).
.........................
http://www.google.com/patents/EP2593439A2?cl=en
Example 2. Preparation of l -(3-teri-butyl-5-(6-hydroxynaphthalen-2-yl)-4- methoxyphenyl)pyrimidine-2,4(l f,3H)-dione (compound (4)).

[00165] This reaction is sensitive to oxygen, and so all vessels were sealed with rubber septa. All solution transfers were accomplished by cannula technique using nitrogen as the inert gas. Anhydrous tetrahydrofuran was sparged with nitrogen gas for 2 hours prior to use to render it anaerobic. Hereafter this is referred to as degassed tetrahydrofuran. [00166] A 100-mL round-bottom flask was charged with 12.9 g of potassium phosphate tribasic (60.8 mmol, 2.0 equivalents), a magnetic stir bar, and 60 mL of water. The mixture was stirred to dissolve the solids, and the aqueous solution was sparged with nitrogen gas for 2 hours prior to use. Hereafter this is referred to as the phosphate solution.
[00167] A 100-mL round-bottom flask was purged with nitrogen gas and charged with 282 mg of tris(dibenzylideneacetone)dipalladium(0) (0.31 mmol, 0.02 equivalents Pd), 413 mg ofphosphine ligand, l ,3,5,7-tetramethyl-8-phenyl-2,4,6-trioxa-8-phosphatricyclo[3.3.1.1 '7]decane (1.4 mmol, 2.3 equivalents relative to Pd) and a magnetic stir bar. The flask was sealed with a septum and the atmosphere above the solids was purged with nitrogen gas. Sixty mL of degassed tetrahydrofuran was added to the flask and the mixture was stirred under a nitrogen atmosphere. This solution was sparged for 15 minutes prior to use and is hereafter referred to as the catalyst solution.
[00168] A 500-mL jacketed reactor was equipped with an overhead stirrer and reflux condenser and the atmosphere was purged with nitrogen gas. The reactor was charged with 12.1 g of l -(3-?er?-butyl-5-iodo- 4-methoxyphenyl)pyrimidine-2,4(l f,3 /)-dione, (30.3 mmol, 1.0 equivalent) and 5.98 g of 6- hydroxynaphthalen-2-ylboronic acid (31.8 mmol, 1.05 equivalents). The atmosphere was purged with nitrogen gas with stirring of the solid reagents for 20 minutes. The reactor was charged with 120 mL of degassed tetrahydrofuran, and the mixture was stirred to dissolve the solids. The solution was sparged with nitrogen gas for 10 minutes. The phosphate solution was added to the reactor by cannula, followed by the catalyst solution. The resulting biphasic mixture was stirred aggressively to ensure adequate phase mixing, and the jacket was warmed to 65 °C. The reaction jacket was cooled to room temperature prior to quench.
[00169] After 2.5 hours, the reaction jacket was cooled to room temperature prior to quench.
[00170] The workup of the reaction was also conducted under anaerobic conditions. Fifty-seven grams of sodium chloride and 4.2 g of cysteine (15 weight equivalents relative to palladium catalyst) were dissolved in 300 mL of water, and the resulting solution was sparged for 2 hours prior to use. To quench the reaction, approximately 1/3 of this solution was transferred to the reaction mixture by cannula under nitrogen gas and the resulting biphasic mixture was stirred vigorously for 2 hours. The mechanical agitation was halted, the two solutions were allowed to separate, and the aqueous solution was drained out of the reactor through the bottom valve. Approximately 1/3 of the quench solution was transferred to the reaction mixture by cannula under nitrogen gas and the resulting biphasic mixture was stirred vigorously for 45 minutes. The mechanical agitation was halted, the two solutions were allowed to separate, and the aqueous solution was drained out of the reactor through the bottom valve. The final portion of the quench solution was transferred to the reaction mixture by cannula, the resulting biphasic mixture was stirred vigorously for 45 minutes and the aqueous solution was drained out of the reactor through the bottom valve. [00171] The remainder of the workup was not conducted under anaerobic conditions. The pale yellow organic solution was drained from the reactor through the bottom valve and filtered over a pad of grade 4 Filtrol® (1 cm deep by 4.5 cm diameter). The reactor and filter cake were rinsed with 70 mL of tetrahydrofuran. The bulk of the solvent was distilled in vacuo (ca 90-130 torr) at ca 40 °C with good agitation from an overhead stirrer. The solution was concentrated to approximately 50 mL volume, during which time the product began to precipitate out. Ethyl acetate (100 mL, 8 volume/weight relative to product) was added to the mixture, and the resultant slurry was stirred overnight at room temperature. The crystalline material was isolated by filtration and the filter cake was washed twice with 20 mL portions of ethyl acetate. The wet-cake was air-dried on the filter and dried in a vacuum oven at 50 °C at approximately 250 torr with a gentle nitrogen sweep overnight.
[00172] The desired product was isolated as a white solid (11.6 g, 96.4% potency vs. standard, 88% potency-adjusted yield).!H NMR (400 MHz, DMSO-4) δ ppm δ 1 1.39 (d, J = 2.1 Hz, 1H), 9.82 (s, 1H), 7.91 (d, J = 0.8 Hz, 1H), 7.80 (d, J= 8.9 Hz, 1H), 7.77 - 7.74 (m, 2H), 7.58 (dd, J = 8.5, 1.7 Hz, 1H), 7.32 (d, J = 2.7 Hz, 1H), 7.27 (d, J= 2.7 Hz, 1H), 7.16 (d, J = 2.3 Hz, 1H), 7.10 (dd, J = 8.8, 2.4 Hz, 1H), 5.64 (dd, J = 7.9, 2.2 Hz, 1H), 3.23 (s, 3H), 1.41 (s, 9H).
Example 3. Preparation of 6-(3-?eri-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-l (2H)-yl)-2- methoxyphenyl)naphthalen-2-yl 1 ,1 ,2,2,3,3,4,4,4-nonafluorobutane-l -sulfonate (compound (5a)).

[00174] A reactor was equipped with an overhead stirrer in the central neck and charged with 45.0 g of 1- (3-?eri-butyl-5-(6-hydroxynaphthalen-2-yl)-4-methoxyphenyl)pyrimidine-2,4(l f,3H)-dione (97.8 weight%>, 106 mmol, 1.0 equivalent) and 21.9 g of 325 mesh potassium carbonate (159 mmol, 1.5 equivalents). The atmosphere was purged with nitrogen gas while the solids were stirred. The flask was charged with 445 mL of Λ^Λ^-dimethylformamide, and the slurry was stirred to dissolve the l-(3-?eri- butyl-5-(6-hydroxynaphthalen-2-yl)-4-methoxyphenyl)pyrimidine-2,4(l f,3H)-dione. The purge was stopped and the reaction was conducted under a slight positive pressure of nitrogen gas.
Perfluorobutanesulfonyl fluoride (35.2 g, 117 mmol, 1.1 equivalents) was added in one portion, and the mixture was stirred vigorously to mix the immiscible liquids overnight.
[00175] The inorganic solids were separated by filtration, and the flask and filter cake were rinsed with approximately 30 mL of Λ^,Λ^-dimethylformamide. The Λ^,Λ^-dimethylformamide solution was filtered directly into a second flask with an overhead stirrer. With stirring, 1 12 g of water (25 weight% of total Λ^,Λ^-dimethylformamide employed) was added to the Λ^,Λ^-dimethylformamide solution of product over approximately 0.5 hour to induce precipitation of the desired product, and the mixture was allowed to stir for 5 hours. The wet-cake was isolated by filtration with recirculation of the liquors to recover all the solids. The wet-cake was washed with 60 mL of 25% (v/v) water yV-dimethylformamide, then 85 mL water.
[00176] The solids were dissolved in 760 mL of isopropyl acetate. The resultant organic solution was washed once with 200 mL of water, twice with 270 mL portions of water and once with 200 mL of water to remove residual AyV-dimethylformamide. Solvent was removed by distillation at approximately 130 torr with heating to 55 °C until the total volume was approximately 200 mL. With efficient stirring, heptane (450 mL) was added to the warm (55 °C) slurry. The slurry was allowed to cool to room temperature overnight with stirring. The desired product was isolated by filtration, with recycling of the liquors to isolate all of the solids material. The wet-cake was washed twice with 100 mL portions of 20% (v/v) isopropyl acetate/heptane. The wet-cake was air-dried on the filter and dried in a vacuum oven at 50 °C at approximately 250 torr with a gentle nitrogen sweep overnight. The title compound was isolated as a white solid (64.0 g, 100% potency vs. standard, 87% yield). !H NMR (600 MHz, DMSO- d6) δ ppm 1 1.42 (s, 1H), 8.21 - 8.15 (m, 4H), 7.84 (dd, J = 8.6, 1.7 Hz, 1H), 7.77 (d, J = 7.9 Hz, 1H), 7.60 (dd, J = 9.0, 2.5 Hz, 1H), 7.39 (d, J = 2.7 Hz, 1H), 7.35 (d, J = 2.7 Hz, 1H), 5.66 (d, J = 7.9 Hz, 1H), 3.21 (s, 3H), 1.41 (s, 9H).
[00177] Example 3-1. Alternative Preparation of 6-(3-teri-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin- 1 (2//)-yl)-2-methoxyphenyl)naphthalen-2-yl 1 , 1 ,2,2,3 ,3 ,4,4,4-nonafluorobutane- 1 -sulfonate (compound (5a)).
[00178] A 250-L, 3-neck round-bottom flask equipped with an overhead stirrer was charged with 10 g of 1 -(3-ier?-butyl-5-(6-hydroxynaphthalen-2-yl)-4-methoxyphenyl)pyrimidine-2,4( l//,3//)-dione (98 wt%>, 23.5 mmol, 1.0 equiv) and 6.5 g of milled potassium carbonate (325 mesh, 47.1 mmol, 2.0 equiv). Acetonitrile (MeCN, 60 mL, 6 volumes with respect to naphthol) and dimethylformamide (dimethylformide, 40 mL, 4 volumes with respect to naphthol) was charged to the reactor and the slurry was stirred. Perfluorobutanesulfonyl fluoride (96 wt%>, 8.3 g, 26 mmol, 1.1 equiv) was charged to the well-stirred mixture over 60 minutes by syringe pump. A trace (<0.1 area%) of starting material was detected by HPLC analysis of an aliquot at 20 minutes reaction time. The
acetonitrile/dimethylformamide solution was filtered over a coarse fritted funnel to separate the inorganic solids, and the flask and filter was rinsed with 15 mL of 3 :2 (v/v)
acetonitrile/dimethylformamide. The total mass of solvents employed was approximately 92 g.
[00179] First crystallization: The acetonitrile/dimethylformamide solution was transferred to a 3- neck flask equipped with an overhead stirrer. Water (50 g, 54 wt%> with respect to total solution charged) was added to the well-stirred solution over 100 minutes. This adjusts the solvent
composition to 35 wt% water. During the addition of water the mixture self-seeded, and the solution was held for approximately 1 hour after complete addition of water. The solids were isolated by filtration, and the wetcake was washed with two 30 mL portions of a rinse solution of 40 wt%
water/27 wt% dimethylformamide/33 wt% acetonitrile and then once with 40 mL of water.
[00180] Aqueous washing: A 500-L jacketed cylindrical reactor equipped with an overhead stirrer and Teflon baffle to aid in vertical mixing was charged with the wetcake and 133 g of ethyl acetate (8X theoretical mass of product, 150 mL). The mixture was stirred to dissolve the substrate and the solution was washed twice with 40 mL portions of water.
[00181] Concentration and crystallization: A constant-volume distillation was conducted with heptanes, in vacuo (ca 100 mmHg, jacket temperature of 50 °C), to adjust the solvent composition to approximately 12 wt% ethyl acetate/88 wt% heptanes. During the distillation, solids begin to crystallize out of the solution. Once the distillation was complete, the solution was cooled to ambient temperature (23 °C). The solids were isolated by filtration and the wet cake was washed with a 50-mL portions heptane. The wet cake was dried to give the final product (14.0 g). The solids were 98.1% pure by HPLC analysis and 100% potent vs. reference standard, for an isolated yield of 85%o.
[00182] Example 4. Preparation of ^-(6-(3-ier?-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-l(2H)-yl)-2- methoxyphenyl)naphthalen-2-yl)methanesulfonamide (compound (A)).

[00183] A 3-L, 3-neck, round-bottom flask was equipped with an overhead stirrer, a thermocouple, a Claisen condenser and a reflux condenser. Tris(dibenzylideneacetone)dipalladium(0) (0.330 g, 0.360 mmol), di-ier?-butyl(2',4',6'-triisopropyl-3,4,5,6-tetramethylbiphenyl-2-yl)phosphine (0.416 g, 0.864 mmol) and milled potassium phosphate tribasic (21.0 g, 99.0 mmol) were charged to the 3-L flask. The flask was purged with argon for not less than 90 minutes with constant stirring of the solids. i-Amyl alcohol (250 ml) was purged with argon for not less than 30 minutes and was transferred to the 3-L flask using a cannula under argon atmosphere. The contents of the 3-L flask were heated to 80 °C and stirred at this temperature for 30 minutes. A 1-L round bottom flask equipped with a magnetic stir bar was charged with 6-(3-ier?-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-l(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl 1,1,2,2,3,3,4,4,4-nonafluorobutane-l-sulfonate (62.9 g, 90 mmol), methanesulfonamide (12.85 g, 135 mmol) and i-amyl alcohol (505 mL), purged with argon and heated to 60 °C. The reaction mixture was stirred under argon for not less than 30 minutes. A clear yellow solution was observed. This solution was transferred to the 3-L flask using a cannula under argon atmosphere. The temperature of the 3-L flask was raised to 85 °C and the contents were stirred for 14 hours under a positive pressure of argon. The temperature was then raised to 95 °C and the contents were stirred for an additional 4 hours under a positive pressure of argon. The reaction mixture was allowed to cool down to room temperature, diluted with tetrahydrofuran (2200 mL) and water (800 mL) and was transferred to a 6-L separatory funnel. The organic layer was washed thrice with water (2000 mL) containing L-cysteine (17.3 g) and NaCl (235 g). The organic layer was collected, filtered through a pad of diatomaceous earth and was concentrated in vacuo to approximately 250 mL. Ethyl acetate (775 mL) was added over 7 hours with stirring, and the mixture was allowed to stir for an additional 14 hours. White solid was isolated by filtration, and the solid was washed with ethyl acetate (1000 mL). The solid was dissolved in tetrahydrofuran (1500 mL) and filtered through a pad of diatomaceous earth to obtain a clear solution. The diatomaceous earth was washed with tetrahydrofuran (300 mL). The combined tetrahydrofuran solution was concentrated in vacuo to approximately 250 mL, and then ethyl acetate (775 mL) was added over 7 hours with stirring. The product solution was allowed to stir for an additional 14 hours. White solid was isolated by filtration. The solid was washed with ethyl acetate (1000 mL) and dried in a vacuum oven at 60 °C for 24 hours. The solid was slurried in 308 mL of 200 proof ethanol for 1.5 hours, then isolated by filtration. The solid was washed with 132 mL of 200 proof ethanol and dried in a vacuum oven at 50 °C for 18 hours. The title compound was isolated as a white solid (32.6 g, 100% potency vs. standard, 73% yield). !H NMR (400 MHz, DMSO-i/6) δ ppm 11.41 (d, J = 2.1 Hz, 1H), 10.04 (s, 1H), 8.02 (d, J = 0.9 Hz, 1H), 7.98 - 7.91 (m, 2H), 7.79 (d, J = 7.9 Hz, 1H), 7.72 (d, J = 2.0 Hz, 1H), 7.69 (dd, J = 8.5, 1.7 Hz, 1H), 7.41 (dd, J = 8.8, 2.2 Hz, 1H), 7.36 (d, J = 2.7 Hz, 1H), 7.31 (d, J = 2.7 Hz, 1H), 5.65 (dd, J = 7.9, 2.2 Hz, 1H), 3.24 (s, 3H), 3.08 (s, 3H), 1.42 (s, 9H).
[00184] Other ligands such as 2,2,7,7-tetramethyl-l-(2',4',6'-triisopropylbiphenyl-2-yl)phosphepane; 7,7,9,9-tetramethyl-8-(2',4',6'-triisopropyl-3,6-dimethoxybiphenyl-2-yl)-l,4-dioxa-8- phosphaspiro[4.5]decane; and 8-(2-(2-methoxynaphthalen- 1 -yl)phenyl)-7,7,9,9-tetramethyl- 1 ,4-dioxa-8- phosphaspiro[4.5]decane were tested under the conditions described above and produced favorable yields of greater than 50% of the sulfonamidated product.
PREPN OF SODIUM SALT
Example 5. Preparation of the sodium salt of V-(6-(3-teri-butyl-5-(2,4-dioxo-3,4- dihydropyrimidin- 1 (2H)-yl)-2-methoxyphenyl)naphthalen-2-yl)methanesulfonamide (compound (As)).

[00192] A solution of 2-propanol and water was prepared by combining 18.5 g of water and 512 g of 2- propanol. Hereafter, this solution is referred to as the "antisolvent solution."
[00193] A solution of 2-propanol and water was prepared by combining 23.94 g of water and 564 g of 2- propanol. This solution was cooled in a refrigerator prior to use. Hereafter, this solution is referred to as the "chilled wash solution."
[00194] A jacketed reactor was equipped with an overhead stirrer and charged with 32.0 g (64.8 mmol) of A^-(6-(3-?er^butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-l(2H)-yl)-2-methoxyphenyl)naphthalen-2- yl)methanesulfonamide and 105.9 g of dimethyl sulfoxide. With stirring the mixture was heated to an internal temperature of 68 °C. A solution of 2.66 g of sodium hydroxide (66.5 mmol, 1.026 equiv) in 16 g of water was added to the reactor over several minutes, followed by 12.4 g of 2-propanol while maintaining the internal temperature at 68 °C. Antisolvent solution (24.5 g) was added to the reactor while maintaining the internal temperature at 68 °C. A slurry of 0.32 g of seed crystals of the final product in 22.8 g of antisolvent solution was added to the reactor, followed by a 2.6 g rinse of the flask with antisolvent solution. The reaction mixture was stirred for 1.5 hours while maintaining the internal temperature at 68 °C. Antisolvent solution (354 g) was added to the reactor over 7 hours while maintaining the internal temperature at 68 °C. The contents of the reactor were cooled to an internal temperature of 0 °C over 7 hours and then mixed at 0 °C for 7 hours. The solids were isolated by filtration and washed with 252 g of the chilled wash solution. The isolated solids were dried in a vacuum oven at 50 °C for 19 hours. The title compound was isolated as a white solid (30.7 g, 92% potency vs. free acid standard, 57.2 mmol free acid equivalent, 88% yield). !H NMR (400 MHz, DMSO-i¾) δ ppm 7.75 (s, 1H), 7.72 (d, J= 7.8 Hz, 1H), 7.59 (dd, J= 8.8, 2.2 Hz, 2H), 7.45 (dd, J= 8.5, 1.8 Hz, 1H), 7.27 (d, J = 2.6 Hz, 2H), 7.21 (d, J= 2.7 Hz, 1H), 7.06 (dd, J= 8.8, 2.2 Hz, 1H), 5.62 (d, J= 7.8 Hz, 1H), 3.24 (s, 3H), 2.68 (s, 3H), 1.40 (s, 9H).
......................................
http://www.google.com/patents/US20130224149
Example 2 Preparation of 1-(3-tert-butyl-5-(6-hydroxynaphthalen-2-yl)-4-methoxyphenyl)pyrimidine-2,4(1H,3H)-dione (compound (4a))

This reaction is sensitive to oxygen, and care was taken to establish and maintain an inert atmosphere in the handling and use of air-sensitive materials or mixtures. All solution transfers were accomplished by cannula technique using nitrogen as the inert gas. Anhydrous tetrahydrofuran was sparged with nitrogen gas for 2 hours prior to use to render it anaerobic. Hereafter this is referred to as degassed tetrahydrofuran.
A 100-mL round-bottom flask was charged with 12.9 g of potassium phosphate tribasic (60.8 mmol, 2.0 equivalents), a magnetic stir bar, and 60 mL of water. The mixture was stirred to dissolve the solids, and the aqueous solution was sparged with nitrogen gas for 2 hours prior to use. Hereafter this is referred to as the phosphate solution.
A 100-mL round-bottom flask was purged with nitrogen gas and charged with 282 mg of tris(dibenzylideneacetone)dipalladium(0) (0.31 mmol, 0.02 equivalents Pd), 413 mg of phosphine ligand, 1,3,5,7-tetramethyl-8-phenyl-2,4,6-trioxa-8-phosphatricyclo[3.3.1.13,7]decane (1.4 mmol, 2.3 equivalents relative to Pd) and a magnetic stir bar. The flask was sealed with a septum and the atmosphere above the solids was purged with nitrogen gas. Sixty mL of degassed tetrahydrofuran was added to the flask and the mixture was stirred under a nitrogen atmosphere. This solution was sparged with nitrogen for 15 minutes prior to use and is hereafter referred to as the catalyst solution.
A 500-mL jacketed reactor was equipped with an overhead stirrer and reflux condenser and the atmosphere was purged with nitrogen gas. The reactor was charged with 12.1 g of 1-(3-tert-butyl-5-iodo-4-methoxyphenyl)pyrimidine-2,4(1H,3H)-dione, (30.3 mmol, 1.0 equivalent) and 5.98 g of 6-hydroxynaphthalen-2-ylboronic acid (31.8 mmol, 1.05 equivalents). The atmosphere was purged with nitrogen gas with stirring of the solid reagents for 20 minutes. The reactor was charged with 120 mL of degassed tetrahydrofuran, and the mixture was stirred to dissolve the solids. The solution was sparged with nitrogen gas for 10 minutes. The phosphate solution was added to the reactor by cannula, followed by the catalyst solution. The resulting biphasic mixture was stirred aggressively to ensure adequate phase mixing, and the jacket was warmed to 65° C. The reaction jacket was cooled to room temperature prior to quench.
After 2.5 hours, the reaction jacket was cooled to room temperature prior to quench.
The workup of the reaction was also conducted under anaerobic conditions. Fifty-seven grams of sodium chloride and 4.2 g of cysteine (15 weight equivalents relative to palladium catalyst) were dissolved in 300 mL of water, and the resulting solution was sparged with inert gas for 2 hours prior to use. To quench the reaction, approximately ⅓ of this solution was transferred to the reaction mixture by cannula under nitrogen gas and the resulting biphasic mixture was stirred vigorously for 2 hours. The mechanical agitation was halted, the two solutions were allowed to separate, and the aqueous solution was drained out of the reactor through the bottom valve. Approximately ⅓ of the quench solution was transferred to the reaction mixture by cannula under nitrogen gas and the resulting biphasic mixture was stirred vigorously for 45 minutes. The mechanical agitation was halted, the two solutions were allowed to separate, and the aqueous solution was drained out of the reactor through the bottom valve. The final portion of the quench solution was transferred to the reaction mixture by cannula, the resulting biphasic mixture was stirred vigorously for 45 minutes and the aqueous solution was drained out of the reactor through the bottom valve.
The remainder of the workup was not conducted under anaerobic conditions. The pale yellow organic solution was drained from the reactor through the bottom valve and filtered over a pad of grade 4 Filtrol® (1 cm deep by 4.5 cm diameter). The reactor and filter cake were rinsed with 70 mL of tetrahydrofuran. The bulk of the solvent was distilled in vacuo (ca 90-130 torr) at ca 40° C. with good agitation from an overhead stirrer. The solution was concentrated to approximately 50 mL volume, during which time the product began to precipitate out. Ethyl acetate (100 mL, about 8 mL of solvent per gram of the product) was added to the mixture, and the resultant slurry was stirred overnight at room temperature. The crystalline material was isolated by filtration and the filter cake was washed twice with 20 mL portions of ethyl acetate. The wet cake was air-dried on the filter and dried in a vacuum oven at 50° C. at approximately 250 torr with a gentle nitrogen sweep overnight.
The desired product was isolated as a white solid (11.6 g, 96.4% potency vs. standard, 88% potency-adjusted yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 11.39 (d, J=2.1 Hz, 1H), 9.82 (s, 1H), 7.91 (d, J=0.8 Hz, 1H), 7.80 (d, J=8.9 Hz, 1H), 7.77-7.74 (m, 2H), 7.58 (dd, J=8.5, 1.7 Hz, 1H), 7.32 (d, J=2.7 Hz, 1H), 7.27 (d, J=2.7 Hz, 1H), 7.16 (d, J=2.3 Hz, 1H), 7.10 (dd, J=8.8, 2.4 Hz, 1H), 5.64 (dd, J=7.9, 2.2 Hz, 1H), 3.23 (s, 3H), 1.41 (s, 9H).
Example 2-1 Alternative preparation of 1-(3-tert-butyl-5-(6-hydroxynaphthalen-2-yl)-4-methoxyphenyl)pyrimidine-2,4(1H,3H)-dione (compound (4a))
This reaction is air-sensitive and the reaction was conducted under anaerobic conditions. A 100-mL round-bottom flask was purged with nitrogen gas and charged with 229 mg of tris(dibenzylideneacetone)dipalladium(0) (0.25 mmol, 0.02 equivalents Pd), 323 mg of 1,3,5,7-tetramethyl-8-phenyl-2,4,6-trioxa-8-phosphatricyclo[3.3.1.13,7]decane (1.13 mmol, 0.045 equivalents) and a magnetic stir bar. The flask was sealed with a septum and the atmosphere above the solids was purged with nitrogen gas. Sixty mL of degassed tetrahydrofuran was added to the flask and the mixture was stirred under a nitrogen atmosphere for 20 minutes. This solution is hereafter referred to as the catalyst solution.
A 500-mL jacketed reactor was equipped with an overhead stirrer and reflux condenser and the atmosphere was purged with nitrogen gas. The reactor was charged with 10.0 g of 1-(3-tert-butyl-5-iodo-4-methoxyphenyl)pyrimidine-2,4(1H,3H)-dione, (25.1 mmol, 1.0 equivalent), 4.98 g of 6-hydroxynaphthalen-2-ylboronic acid (26.6 mmol, 1.06 equivalents) and 10.3 g of potassium phosphate tribasic (48.7 mmol, 2.0 equivalents). The atmosphere was purged with nitrogen gas with stirring of the solid reagents for 20 minutes. The reactor was charged with 100 mL of tetrahydrofuran, 50 mL of water, and the mixture was stirred to dissolve the solids. The biphasic mixture was sparged with nitrogen gas for 30 minutes. The catalyst solution was transferred to the main reactor by positive nitrogen pressure through a cannula. The resulting biphasic mixture was stirred aggressively and warmed to an internal temperature between 60 and 65° C. under nitrogen for 2 hours. The reaction mixture was cooled to an internal temperature between 50 and 55° C. before quench.
The workup of the reaction was conducted under anaerobic conditions at an internal temperature between 50 and 55° C. Fifteen grams of sodium chloride and 1.0 g of cysteine were dissolved in 80 mL of water, and the resulting solution was sparged for 1 hour. This solution was transferred to the reaction mixture by cannula with nitrogen gas pressure and the resulting biphasic mixture was stirred vigorously for 45 minutes. The mechanical agitation was halted, the two solutions were allowed to separate, and the aqueous solution was drained out of the reactor through the bottom valve. Fifteen grams of sodium chloride and 1.0 g of cysteine were dissolved in 80 mL of water, and the resulting solution was sparged for 1 hour. This solution was transferred to the reaction mixture by cannula with nitrogen gas pressure and the resulting biphasic mixture was stirred vigorously for 45 minutes. The mechanical agitation was halted, the two solutions were allowed to separate, and the aqueous solution was drained out of the reactor through the bottom valve.
The pale yellow organic solution was drained from the reactor through the bottom valve and filtered over a polypropylene filter to remove palladium black. The reactor and filter cake were rinsed with 22 mL of tetrahydrofuran and 50 mL of ethyl acetate was added to the organic solution. The solution was distilled at atmospheric pressure (approximately 66° C. internal temperature) with continuous addition of 110 mL of ethyl acetate, keeping the volume of the solution roughly constant during the distillation. During the constant-volume distillation, solids began to precipitate in the reactor. After the ethyl acetate was charged, the distillation was continued at atmospheric pressure, concentrating the slurry to approximately 60 mL total volume. The solution was cooled to an internal temperature of approximately 30° C. and held for 3 hours with stirring. The crystalline material was isolated by filtration and the filter cake was washed twice with 20 mL portions of ethyl acetate. The wet cake was dried in a vacuum oven at 50° C. with a gentle nitrogen sweep overnight. The desired product was isolated as an off-white solid (8.33 g, 80% yield). 1H NMR (400 MHz, DMSO-d6) δ ppm δ 11.39 (d, J=2.1 Hz, 1H), 9.82 (s, 1H), 7.91 (d, J=0.8 Hz, 1H), 7.80 (d, J=8.9 Hz, 1H), 7.77-7.74 (m, 2H), 7.58 (dd, J=8.5, 1.7 Hz, 1H), 7.32 (d, J=2.7 Hz, 1H), 7.27 (d, J=2.7 Hz, 1H), 7.16 (d, J=2.3 Hz, 1H), 7.10 (dd, J=8.8, 2.4 Hz, 1H), 5.64 (dd, J=7.9, 2.2 Hz, 1H), 3.23 (s, 3H), 1.41 (s, 9H).
Example 2-2 Alternative preparation of 1-(3-tert-butyl-5-(6-hydroxynaphthalen-2-yl)-4-methoxyphenyl)pyrimidine-2,4(1H,3H)-dione (compound (4a))
This reaction is air-sensitive and the reaction was conducted under nitrogen atmosphere. A 100-mL round-bottom flask was purged with nitrogen gas and charged with 303 mg of tris(dibenzylideneacetone)dipalladium(0) (0.33 mmol, 0.02 equivalents Pd), 411 mg of 1,3,5,7-tetramethyl-8-phenyl-2,4,6-trioxa-8-phosphatricyclo[3.3.1.13,7]decane (1.40 mmol, 0.045 equivalents) and a magnetic stir bar. The flask was sealed with a septum and the atmosphere above the solids was purged with nitrogen gas. Seventy-five (75) mL of degassed tetrahydrofuran was added to the flask and the mixture was stirred under a nitrogen atmosphere for 25 minutes. This solution is hereafter referred to as the catalyst solution.
A 500-mL jacketed reactor was equipped with an overhead stirrer and reflux condenser and the atmosphere was purged with nitrogen gas. The reactor was charged with 12.5 g of 1-(3-tert-butyl-5-iodo-4-methoxyphenyl)pyrimidine-2,4(1H,3H)-dione, (31.2 mmol, 1.0 equivalent), 6.20 g of 6-hydroxynaphthalen-2-ylboronic acid (33.0 mmol, 1.06 equivalents) and 13.0 g of potassium phosphate tribasic (61.2 mmol, 2.0 equivalents). The reactor was charged with 130 mL of tetrahydrofuran, 65 mL of water, and the mixture was stirred to dissolve the solids. The biphasic mixture was sparged with nitrogen gas for 30 minutes. The catalyst solution was transferred to the main reactor by positive nitrogen pressure through a cannula. The resulting biphasic mixture was stirred aggressively and warmed to an internal temperature between 60 and 65° C. under nitrogen for 2.5 hours. The reaction mixture was cooled to an internal temperature between 50 and 55° C. before quench.
The workup of the reaction was conducted under anaerobic conditions at an internal temperature between 50 and 55° C. Sodium chloride (18.8 g) and cysteine (1.25 g) were dissolved in 100 mL of water, and the resulting solution was sparged with nitrogen for 40 minutes. This solution was transferred to the reaction mixture by cannula with nitrogen gas pressure and the resulting biphasic mixture was stirred vigorously for 45 minutes. The mechanical agitation was halted, the two solutions were allowed to separate, and the aqueous solution was drained out of the reactor through the bottom valve. Sixty-three (63) mL of degassed tetrahydrofuran were added to the reactor by cannula with positive nitrogen pressure. Sodium chloride (18.9 g) and cysteine (1.333 g) were dissolved in 100 mL of water, and the resulting solution was sparged with nitrogen for 40 minutes. This solution was transferred to the reaction mixture by cannula with nitrogen gas pressure and the resulting biphasic mixture was stirred vigorously for 45 minutes. The mechanical agitation was halted, the two solutions were allowed to separate, and the aqueous solution was drained out of the reactor through the bottom valve.
The pale yellow organic solution was drained from the reactor through the bottom valve and filtered through a thin pad of filter aid on a polyethylene filter while warm. The reactor and filter cake were rinsed with 32 mL of tetrahydrofuran, and 65 mL of ethyl acetate was added to the organic solution. The solution was distilled at atmospheric pressure (approximately 66° C. internal temperature) with continuous addition of 190 mL of ethyl acetate, keeping the volume of the solution roughly constant during the distillation. During the constant-volume distillation, solids began to precipitate in the reactor. After the ethyl acetate was charged, the distillation was continued at atmospheric pressure, concentrating the slurry to approximately 90 mL total volume. The slurry was cooled to an internal temperature of approximately 40° C. and was concentrated further in vacuo to a total volume of approximately 50 mL. The slurry was cooled to an internal temperature of 30° C. and held for 16 hours with stirring. The crystalline material was isolated by filtration, and the filter cake was washed twice with 25 mL portions of ethyl acetate. The wet cake was dried in a vacuum oven at 50° C. with a gentle nitrogen sweep overnight. The desired product was isolated as an off-white solid (11.4 g, 99.5% potent vs. standard, 87% potency-adjusted yield).
Example 4 Preparation of N-(6-(3-tert-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl)methanesulfonamide (compound (A-1))

A 3-L, 3-neck, round-bottom flask was equipped with an overhead stirrer, a thermocouple, a Claisen condenser and a reflux condenser. Tris(dibenzylideneacetone)dipalladium(0) (0.330 g, 0.360 mmol), di-tert-butyl(2′,4′,6′-triisopropyl-3,4,5,6-tetramethylbiphenyl-2-yl)phosphine (0.416 g, 0.864 mmol) and milled potassium phosphate tribasic (21.0 g, 99.0 mmol) were charged to the 3-L flask. The flask was purged with argon for not less than 90 minutes with constant stirring of the solids. t-Amyl alcohol (250 ml) was charged to a separate 500-mL round-bottom flask and was purged with argon for not less than 30 minutes and was transferred to the 3-L flask using a cannula under argon atmosphere. The contents of the 3-L flask were heated to 80° C. and stirred at this temperature for 30 minutes. A 1-L round-bottom flask equipped with a magnetic stir bar was charged with 6-(3-tert-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl-1,1,2,2,3,3,4,4,4-nonafluorobutane-1-sulfonate (62.9 g, 90 mmol), methanesulfonamide (12.85 g, 135 mmol) and t-amyl alcohol (505 mL), purged with argon and heated to 60° C. The reagent mixture was stirred under argon for not less than 30 minutes. A clear yellow solution was observed. This solution was transferred to the 3-L flask using a cannula under argon atmosphere. The temperature of the 3-L flask was raised to 85° C. and the contents were stirred for 14 hours under a positive pressure of argon. The temperature was then raised to 95° C. and the contents were stirred for an additional 4 hours under a positive pressure of argon. The reaction mixture was allowed to cool down to room temperature, diluted with tetrahydrofuran (2200 mL) and water (800 mL) and was transferred to a 6-L separatory funnel. The organic layer was washed thrice with water (2000 mL) containing L-cysteine (17.3 g) and NaCl (235 g). The organic layer was collected, filtered through a pad of diatomaceous earth and was concentrated in vacuo to approximately 250 mL. Ethyl acetate (775 mL) was added over 7 hours with stirring, and the mixture was allowed to stir for an additional 14 hours. White solid was isolated by filtration, and the solid was washed with ethyl acetate (1000 mL). The solid was then dissolved in tetrahydrofuran (1500 mL) and filtered through a pad of diatomaceous earth to obtain a clear solution. The diatomaceous earth was washed with tetrahydrofuran (300 mL). The combined tetrahydrofuran solution was concentrated in vacuo to approximately 250 mL, and then ethyl acetate (775 mL) was added over 7 hours with stirring. The product solution was allowed to stir for an additional 14 hours. White solid was isolated by filtration. The solid was washed with ethyl acetate (1000 mL) and dried in a vacuum oven at 60° C. for 24 hours. The solid was slurried in 308 mL of 200 proof ethanol for 1.5 hours, then isolated by filtration. The solid was washed with 132 mL of 200 proof ethanol and dried in a vacuum oven at 50° C. for 18 hours. The title compound was isolated as a white solid (32.6 g, 100% potency vs. standard, 73% yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 11.41 (d, J=2.1 Hz, 1H), 10.04 (s, 1H), 8.02 (d, J=0.9 Hz, 1H), 7.98-7.91 (m, 2H), 7.79 (d, J=7.9 Hz, 1H), 7.72 (d, J=2.0 Hz, 1H), 7.69 (dd, J=8.5, 1.7 Hz, 1H), 7.41 (dd, J=8.8, 2.2 Hz, 1H), 7.36 (d, J=2.7 Hz, 1H), 7.31 (d, J=2.7 Hz, 1H), 5.65 (dd, J=7.9, 2.2 Hz, 1H), 3.24 (s, 3H), 3.08 (s, 3H), 1.42 (s, 9H). 13C NMR (101 MHz, DMSO-d6) δ ppm 163.1 (C), 156.0 (C), 150.0 (C), 145.3 (CH), 142.9 (C), 136.0 (C), 134.3 (C), 134.2 CO, 133.5 (C), 132.2 (C), 129.5 (C), 129.0 (CH), 127.6 (CH), 127.1 (CH), 127.0 (CH), 126.5 (CH), 124.3 (CH), 120.2 (CH), 114.5 (CH), 101.1 (CH), 60.3 (CH3), 39.4 (CH3), 35.1 (C), 30.5 (CH3).
Other ligands such as 2,2,7,7-tetramethyl-1-(2′,4′,6′-triisopropylbiphenyl-2-yl)phosphepane; 7,7,9,9-tetramethyl-8-(2′,4′,6′-triisopropyl-3,6-dimethoxybiphenyl-2-yl)-1,4-dioxa-8-phosphaspiro[4.5]decane; and 8-(2-(2-methoxynaphthalen-1-yl)phenyl)-7,7,9,9-tetramethyl-1,4-dioxa-8-phosphaspiro[4.5]decane were tested under the conditions described above and produced favorable yields of greater than 50% of the sulfonamidated product.
Example 4-1 Alternative Preparation of N-(6-(3-tert-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl)methanesulfonamide (compound (A-1))
A 450-mL, stainless steel Parr® pressure reactor equipped with an overhead stirrer was charged with tris(dibenzylideneacetone)dipalladium(0) (0.131 g, 0.143 mmol), di-tert-butyl(2′,4′,6′-triisopropyl-3,6-dimethoxybiphenyl-2-yl)phosphine (0.167 g, 0.344 mmol) and milled potassium phosphate tribasic (6.69 g, 31.5 mmol). The flask was purged with argon for not less than 90 minutes. Tetrahydrofuran (90 mL) was taken in a 100-mL round bottom flask, purged with argon for not less than 30 minutes and was transferred to the 450-mL reactor using a cannula under argon atmosphere. The contents of the 450-mL reactor were heated to 80° C. and stirred at this temperature for 30 minutes. A 250-mL, round-bottom flask equipped with a magnetic stir bar was charged with 6-(3-tert-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl-1,1,2,2,3,3,4,4,4-nonafluorobutane-1-sulfonate (20.0 g, 28.6 mmol), methanesulfonamide (3.27 g, 34.4 mmol) and tetrahydrofuran (160 mL), purged with argon for not less than 45 minutes. A clear yellow solution was observed. This solution was transferred to the 450-mL reactor that has been cooled to the room temperature using a cannula under argon atmosphere. The temperature of the 450-mL reactor was raised to 90° C. and the contents were stirred for 20 hours. The reaction mixture was allowed to cool down to 50° C., diluted with tetrahydrofuran (70 mL) and water (70 mL) containing L-cysteine (0.875 g) and sodium chloride (7.7 g). The contents were stirred for 2 hours at 50° C. The aqueous layer was discarded and the organic layer was filtered through an approximately 2-inch pad of diatomaceous earth and rinsed with tetrahydrofuran (45 mL) to obtain a clear, light yellow solution. The total weight of reaction mixture was 363.43 g. HPLC analysis of the reaction mixture revealed 13.71 g (97%) of the title compound was present in the reaction mixture. A portion of the reaction mixture (50 g) was concentrated to a final volume of 12-14 mL under vacuum. Ethyl acetate (45 mL) was added slowly and the reaction mixture was stirred over night at room temperature to obtain white slurry. Product was collected by filtration, washed with ethyl acetate (7 mL) and dried overnight in a vacuum oven at 50-60° C. to obtain 2.02 g of white solid. Ethanol (14 mL) was added to the solid and stirred overnight at the room temperature. The product was collected by filtration, washed with ethanol (4 mL) and dried overnight in a vacuum oven at 50-60° C. to obtain the title compound (1.79 g, 95.4%).
Example 4-2 Alternative preparation of N-(6-(3-tert-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl)methanesulfonamide (compound (A-1))
A 450-mL, stainless steel Parr® pressure reactor equipped with an overhead stirrer was charged with tris(dibenzylideneacetone)dipalladium(0) (0.105 g, 0.115 mmol), di-tert-butyl(2′,4′,6′-triisopropyl-3,6-dimethoxybiphenyl-2-yl)phosphine (0.133 g, 0.275 mmol) and milled potassium phosphate tribasic (5.35 g, 25.2 mmol). The flask was purged with argon for not less than 90 minutes. 2-Methyltetrahydrofuran (70 mL) was taken in a 100-mL round bottom flask, purged with argon for not less than 30 minutes and was transferred to the 450-mL reactor using a cannula under argon atmosphere. The contents of the 450-mL reactor were heated to 80° C. and stirred at this temperature for 30 minutes. A 250-mL, round bottom flask equipped with a magnetic stir bar was charged with 6-(3-tert-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl-1,1,2,2,3,3,4,4,4-nonafluorobutane-1-sulfonate (16.0 g, 22.9 mmol), methanesulfonamide (2.61 g, 27.5 mmol) and 2-methyltetrahydrofuran (155 mL), purged with argon for not less than 60 minutes. This solution was transferred to the 450-mL reactor that has been cooled to the room temperature using a cannula under argon atmosphere. The temperature of the 450-mL flask was raised to 90° C. and the contents were stirred for 14 hours. The reaction mixture was allowed to cool down to 70° C., diluted with ethyl acetate (190 mL) and stirred for 3 hours at 70° C., cooled to the room temperature, stirred for an additional 4 hours, filtered through a fine frit filter funnel and rinsed with ethyl acetate (90 mL) to obtain 29.4 g of light brown solid. A portion of this solid (13.04 g) was transferred to a 500-mL, 3-neck round bottom flask equipped with an overhead stirrer and a thermocouple. Tetrahydrofuran (175 mL) was added, followed by the addition of water 50 mL containing L-cysteine (0.63 g) and sodium chloride (5.5 g). The reaction mixture was stirred for 2 hours at 50° C. under a slight positive pressure of argon. The reaction mixture was transferred to a 500-mL separatory funnel and the aqueous layer was discarded. The organic layer was filtered through an approximately 2-inch pad of diatomaceous earth and rinsed with tetrahydrofuran (45 mL) to obtain a clear, light yellow solution. The organic layer was concentrated to a total weight of 45.59 g. A portion of this organic solution (41.58 g) was charged to a 250-mL, 3-neck round bottom flask fitted with an overhead stirrer. Ethyl acetate (80 mL) was added over 6 hours by a pump with constant stirring at room temperature. The product was collected by filtration, rinsed with ethyl acetate (20 mL) and dried in a vacuum oven for 2 hours to obtain 3.17 g of the title compound (>99.8 pure and 94.6% potent vs. standard).
Example 4-3 Alternative preparation of N-(6-(3-tert-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl)methanesulfonamide (compound (A-1))
A 600-mL, stainless steel Parr® pressure reactor equipped with an overhead stirrer was charged with tris(dibenzylideneacetone)dipalladium(0) (0.229 g, 0.251 mmol), di-tert-butyl(2′,4′,6′-triisopropyl-3,6-dimethoxybiphenyl-2-yl)phosphine (0.291 g, 0.601 mmol) and milled potassium phosphate tribasic (11.70 g, 55.1 mmol). The flask was purged with argon for not less than 90 minutes. Ethyl acetate (140 mL) was taken in a 250-mL, round bottom flask, purged with argon for not less than 30 minutes and was transferred to the 600-mL reactor using a cannula under argon atmosphere. The contents of the 600-mL reactor were heated to 80° C. and stirred at this temperature for 30 minutes. A 500-mL round bottom flask equipped with a magnetic stir bar was charged with 6-(3-tert-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl-1,1,2,2,3,3,4,4,4-nonafluorobutane-1-sulfonate (35.0 g, 50.1 mmol), methanesulfonamide (5.72 g, 60.1 mmol) and ethyl acetate (280 mL), purged with argon for not less than 60 minutes while stirring at 50° C. This solution was transferred to the 600-mL reactor that had been cooled to room temperature using a cannula under argon atmosphere. The temperature of the 600-mL flask was raised to 90° C., and the contents were stirred for 18 hours. The reaction mixture was allowed to cool down to 40° C., filtered and rinsed with ethyl acetate (140 mL). Solid (41.50 g) was obtained after drying for 2 hours on high vacuum. This solid contained the titled product (23.06 g, 93%).
Example 4-4 Alternative preparation of N-(6-(3-tert-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl)methanesulfonamide (compound (A-1))
Tris(dibenzylideneacetone)dipalladium(0) (0.0066 g, 7.16 μmol), di-tert-butyl(2′,4′,6′-triisopropyl-3,6-dimethoxybiphenyl-2-yl)phosphine (0.0083 g, 17 μmol) and milled potassium phosphate tribasic (0.334 g, 1.58 mmol) were charged to a 40-mL reaction vial inside an inert atmosphere glove box. t-Amyl alcohol (4 mL) was added, the vial was capped, and the contents were heated to 80° C. and stirred at this temperature for 30 minutes. The reaction mixture was cooled down to the room temperature. 6-(3-tert-Butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl-1,1,2,2,3,3,4,4,4-nonafluorobutane-1-sulfonate (1.0 g, 1.43 mmol), methanesulfonamide (0.163 g, 1.72 mmol) and t-amyl alcohol (8 mL) were added to the 40-mL reaction vial, and the vial was capped. The reaction temperature was raised to 90° C. and the contents were stirred for 5 hours. HPLC analysis of the reaction mixture showed that the product was formed in 94 area % at 210 nm.
Example 4-5 Alternative preparation of N-(6-(3-tert-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl)methanesulfonamide (compound (A-1))
A 600-mL, stainless steel, Parr® reactor was equipped with an overhead stirrer, thermocouple and a heating mantle. Tris(dibenzylideneacetone)dipalladium(0) (0.164 g, 0.179 mmol), di-tert-butyl(2′,4′,6′-triisopropyl-3,6-dimethoxybiphenyl-2-yl)phosphine (0.208 g, 0.429 mmol) and milled potassium phosphate tribasic (8.36 g, 39.4 mmol) were charged to the 600-mL reactor. The reactor was purged with argon for not less than 90 minutes. 2-Methyltetrahydrofuran (100 mL) was purged with argon for not less than 30 minutes and was transferred to the 600-mL reactor using a cannula under argon atmosphere. The reactor was tightly sealed, the contents were heated to 80° C. and stirred at this temperature for 30 minutes. A 500-mL round bottom flask equipped with a magnetic stir bar was charged with 6-(3-tert-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl 1,1,2,2,3,3,4,4,4-nonafluorobutane-1-sulfonate (25 g, 35.8 mmol), methanesulfonamide (4.09 g, 42.9 mmol) and ethyl acetate (200 mL), purged with argon for not less than 30 minutes with stirring and heated to 60° C. A clear solution was observed. This solution was transferred to the 600-mL reactor using a cannula under argon atmosphere. The reactor was tightly sealed, the contents were heated to 90° C. and stirred at this temperature for 14 hours. The reaction mixture was cooled to 35° C., solids were collected by filtration, washed with ethyl acetate (300 mL) and dried under high vacuum for 2-4 hours. The solids were then transferred to a 1-L, three-neck, round-bottom flask equipped with an overhead stirrer and a thermocouple. N-Acetyl-L-cysteine (0.58 g, 3.5 mmol), dimethylformamide (DMF) (100 mL) and glacial acetic acid (0.85 g) were charged to the 1-L flask; the contents were heated to 60° C. and mixed for 1 hour. The mixture was filtered through approximately 2-inch pad of diatomaceous earth and washed with DMF (50 mL). The dark-brown/black-colored solid collected on diatomaceous earth was discarded and the light yellow/clear filtrate was charged to a separate 1-L, three-neck, round-bottom flask equipped with an overhead stirrer, a thermocouple and a syringe pump. The DMF solution was mixed and methanol (300 mL) was added over 8 hours, while maintaining the internal temperature at 25±5° C. The white solid was collected by filtration washed with methanol (150 mL) and dried in a vacuum oven at 50° C. for not less than 8 hours. The title compound was isolated as a white solid (15.8 g, 89% yield).
Example 4-6 Alternative preparation of N-(6-(3-tert-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl)methanesulfonamide (compound (A-1))
A 600-mL, stainless steel, Parr® reactor was equipped with an overhead stirrer, thermocouple and a heating mantle. Tris(dibenzylideneacetone)dipalladium(0) (0.164 g, 0.179 mmol), 7,7,9,9-tetramethyl-8-(2′,4′,6′-triisopropyl-3,6-dimethoxybiphenyl-2-yl)-1,4-dioxa-8-phosphaspiro[4.5]decane (0.238 g, 0.429 mmol) and milled potassium phosphate tribasic (8.36 g, 39.4 mmol) were charged to the 600-mL reactor. The reactor was purged with argon for not less than 90 minutes. 2-Methyltetrahydrofuran (100 mL) was purged with argon for not less than 30 minutes and was transferred to the 600-mL reactor using a cannula under argon atmosphere. The reactor was tightly sealed, the contents were heated to 80° C. and stirred at this temperature for 30 minutes. A 500-mL round bottom flask equipped with a magnetic stir bar was charged with 6-(3-tert-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl 1,1,2,2,3,3,4,4,4-nonafluorobutane-1-sulfonate (25 g, 35.8 mmol), methanesulfonamide (4.09 g, 42.9 mmol) and ethyl acetate (200 mL), purged with argon for not less than 30 minutes with stirring and heated to 60° C. A clear solution was observed. This solution was transferred to the 600-mL reactor using a cannula under argon atmosphere. The reactor was tightly sealed, the contents were heated to 90° C. and stirred at this temperature for 14 hours. The reaction mixture was cooled to 35° C., 5% aqueous N-acetyl-L-cysteine solution (100 mL) was added and the contents were mixed for 1 hour at 35° C. Solids were collected by filtration, washed with water (2×25 mL) and ethyl acetate (3×80 mL) and were dried under high vacuum for 2-4 hours. The solids were then transferred to a 1-L, three-neck, round-bottom flask equipped with an overhead stirrer and a thermocouple. N-Acetyl-L-cysteine (0.58 g, 3.5 mmol), dimethylformamide (DMF) (100 mL) and glacial acetic acid (0.85 g) were charged to the 1-L flask; the contents were heated to 60° C. and mixed for 1 hour. The mixture was filtered through an approximately 2-inch pad of diatomaceous earth and washed with DMF (50 mL). The dark-brown/black-colored solid collected on the diatomaceous earth was discarded and the light yellow/clear filtrate was charged to a separate 1-L, three-neck, round-bottom flask equipped with an overhead stirrer, a thermocouple and a syringe pump. The DMF solution was mixed and methanol (300 mL) was added over 8 hours, while maintaining the internal temperature at 25±5° C. The white solid was collected by filtration washed with methanol (150 mL) and dried in a vacuum oven at 50° C. for not less than 8 hours. The title compound was isolated as a white solid (15.6 g, 88% yield).
Example 4-7 Alternative preparation of N-(6-(3-tert-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl)methanesulfonamide (compound (A-1))

Tris(dibenzylideneacetone)dipalladium(0) (0.0026 g, 2.80 μmol), di-tert-butyl(2′,4′,6′-triisopropyl-3,6-dimethoxybiphenyl-2-yl)phosphine (0.0033 g, 6.72 μmol) and milled potassium phosphate tribasic (0.131 g, 0.616 mmol) were charged to a 40-mL reaction vial inside an inert atmosphere glove box. 2-Methyltetrahydrofuran (1.5 mL) was added, the vial was capped, and the contents were heated to 80° C. and stirred at this temperature for 30 minutes. The reaction mixture was cooled down to room temperature. 6-(3-tert-Butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl 1,1,2,2-tetrafluoro-2-(perfluoroethoxy)ethanesulfonate (0.4 g, 0.560 mmol, Example 3-7, compound (5f)), methanesulfonamide (0.064 g, 0.672 mmol) and ethyl acetate (3 mL) were added to the 40-mL reaction vial. The temperature of the closed vial was raised to 90° C. and the contents were magnetically stirred for 16 hours. HPLC analysis of the reaction mixture showed that the product was formed in 97 area % at 210 nm.
Example 4-8 Alternative preparation of N-(6-(3-tert-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl)methanesulfonamide (compound (A-1))

Tris(dibenzylideneacetone)dipalladium(0) (0.0071 g, 7.71 μmol), di-tert-butyl(2′,4′,6′-triisopropyl-3,6-dimethoxybiphenyl-2-yl)phosphine (0.0089 g, 19.0 μmol) and milled potassium phosphate tribasic (0.360 g, 1.696 mmol) were charged to a 40-mL reaction vial inside an inert atmosphere glove box. 2-Methyltetrahydrofuran (4 mL) was added, and the closed vial and its contents were heated to 80° C. with magnetic stirring for 30 minutes. The reaction mixture was cooled down to room temperature. 6-(3-tert-Butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl 1,1,1,2,3,3,3-heptafluoropropane-2-sulfonate (1.0 g, 1.542 mmol, Example 3-4, compound (5c)), methanesulfonamide (0.176 g, 1.850 mmol) and ethyl acetate (8 mL) were added to the 40-mL reaction vial. The temperature of the closed vial and its contents was raised to 90° C. and stirred for 20 hours. HPLC analysis of the reaction mixture showed that the product was formed in 95 area % at 210 nm.
Example 4-9 Alternative preparation of N-(6-(3-tert-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl)methanesulfonamide (compound (A-1))

Tris(dibenzylideneacetone)dipalladium(0) (0.0055 g, 6.02 μmol), di-tert-butyl(2′,4′,6′-triisopropyl-3,6-dimethoxybiphenyl-2-yl)phosphine (0.0070 g, 14.0 μmol) and milled potassium phosphate tribasic (0.281 g, 1.324 mmol) were charged to a 40-mL reaction vial inside an inert atmosphere glove box. 2-Methyltetrahydrofuran (3.4 mL) was added, and the closed vial and its contents were heated to 80° C. with magnetic stirring for 30 minutes. The reaction mixture was cooled down to room temperature. 6-(3-tert-Butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl sulfofluoridate (0.6 g, 1.204 mmol, Example 3-8, compound (5g)), methanesulfonamide (0.137 g, 1.444 mmol) and ethyl acetate (6.7 mL) were added to the 40-mL reaction vial. The temperature of the closed reaction vial and its contents was raised to 90° C. and the contents were stirred for 20 hours. HPLC analysis of the reaction mixture showed that the product was formed in 79 area % at 210 nm.
Example 4-10 Alternative preparation of N-(6-(3-tert-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl)methanesulfonamide (compound (A-1))

Tris(dibenzylideneacetone)dipalladium(0) (0.0042 g, 4.56 μmol), di-tert-butyl(2′,4′,6′-triisopropyl-3,6-dimethoxybiphenyl-2-yl)phosphine (0.0053 g, 12.0 μmol) and milled potassium phosphate tribasic (0.213 g, 1.003 mmol) were charged to a 40-mL reaction vial inside an inert atmosphere glove box. 2-Methyltetrahydrofuran (1.9 mL) was added, and the closed vial and its contents were heated to 80° C. with magnetic stirring for 30 minutes. The reaction mixture was cooled down to room temperature. 6-(3-tert-Butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl trifluoromethanesulfonate (0.5 g, 0.912 mmol, Example 3-6, compound (5e)), methanesulfonamide (0.104 g, 1.094 mmol) and ethyl acetate (5.7 mL) were added to the 40-mL reaction vial. The temperature of the closed vial and its contents was raised to 90° C. and stirred for 14 hours. HPLC analysis of the reaction mixture showed that the product was formed in 91 area % at 210 nm.
Example 4-11 Alternative preparation of N-(6-(3-tert-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl)methanesulfonamide (compound (A-1))

Tris(dibenzylideneacetone)dipalladium(0) (0.0037 g, 4.04 μmol), di-tert-butyl(2′,4′,6′-triisopropyl-3,6-dimethoxybiphenyl-2-yl)phosphine (0.0047 g, 9.7 μmol) and milled potassium phosphate tribasic (0.094 g, 0.445 mmol) were charged to a 40-mL reaction vial inside an inert atmosphere glove box. tert-Amyl alcohol (1.0 mL) was added, the contents were heated to 80° C. and stirred at this temperature for 30 minutes. The reaction mixture was cooled down to room temperature. 6-(3-tert-Butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl methanesulfonate (0.2 g, 0.404 mmol), methanesulfonamide (0.046 g, 0.485 mmol) and tert-amyl alcohol (1.5 mL) were added to a 40-mL reaction vial. The reaction temperature was raised to 110° C., and the contents were stirred for 14 hours. HPLC analysis of the reaction mixture showed that the titled compound was formed in 7 area % at 210 nm.
Example 4-12 Alternative preparation of N-(6-(3-tert-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl)methanesulfonamide (compound (A-1))

Palladium acetate (0.0018 g, 8.09 μmol), di-tert-butyl(2′,4′,6′-triisopropyl-3,6-dimethoxybiphenyl-2-yl)phosphine (0.0086 g, 0.018 mmol) and water (0.6 μL, 0.032 mmol) were charged to a 40-mL reaction vial inside an inert atmosphere glove box. tert-Amyl alcohol (1.0 mL) was added, and the contents were heated to 80° C. and stirred at this temperature for 15 minutes. The reaction mixture was cooled down to room temperature. Potassium phosphate tribasic (0.094 g, 0.445 mmol), 6-(3-tert-butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl methanesulfonate (0.2 g, 0.404 mmol), methanesulfonamide (0.046 g, 0.485 mmol) and tert-amyl alcohol (1.5 mL) were added to the 40-mL reaction vial. The reaction temperature was raised to 110° C., and the contents were stirred for 14 hours. HPLC analysis of the reaction mixture showed that the titled compound was formed in 5 area % at 210 nm.
.........................
REF...
Wagner, Rolf et al, Uracil or thymine derivative for treating hepatitis C and their preparation, PCT Int. Appl., WO2009039127, 26 Mar 2009
Flentge, Charles A. et al, Preparation of anti-infective pyrimidines for treating hepatitis C,PCT Int. Appl., WO2009039134, 26 Mar 2009
Shekhar, Shashank et al,N-(6-(3-tert-Butyl-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-2-methoxyphenyl)naphthalen-2-yl)methanesulfonamide as HCV polymerase inhibitor and its preparation, pharmaceutical compositions and use in the treatment of hepatitis C,PCT Int. Appl., WO2012009699, 19 Jan 2012
Shekhar, Shashank et al,Process for preparing antiviral pyrimidinylphenylnaphthalenyl sulfonamide compounds,PCT Int. Appl.,US20130224149, 29 Aug 2013
Shekhar, Shashank et al,Preparation and use of phosphine ligands for catalytic reactions,U.S. Pat. Appl. Publ., US20130217876, 22 Aug 2013
23 IDX 18719; IDX 719; Samatasvir
- IDX 18719; IDX 719; Samatasvir
N-((1R)-2-((2S)-2-(5-(4-(6-(2-((2S)-1-((2S)-2-((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-3H-benzimidazol-5-yl)thieno(3,2-b)thiophen-3-yl)phenyl)-1H-imidazol-2-yl)pyrrolidin-1-yl)-2-oxo-1-phenylethyl)carbamate
Carbamic acid, N-((1R)-2-((2S)-2-(5-(4-(6-(2-((2S)-1-((2S)-2-((methoxycarbonyl)amino)-3-methyl-1-oxobutyl)-2-pyrrolidinyl)-1H-benzimidazol-6-yl)thieno(3,2-b)thien-3-yl)phenyl)-1H-imidazol-2-yl)-1-pyrrolidinyl)-2-oxo-1-phenylethyl)-, methyl ester
Carbamic acid, N-[(1R)-2-[(2S)-2-[5-[4-[6-[2-[(2S)-1-[(2S)-2-[(methoxycarbonyl)amino]-3-methyl-1-oxobutyl]-2-pyrrolidinyl]-1H-benzimidazol-6-yl]thieno[3,2-b]thien-3-yl]phenyl]-1H-imidazol-2-yl]-1-pyrrolidinyl]-2-oxo-1-phenylethyl]-, methyl ester
[(5)-l-((5)-2- {6-[5-(4- {(5)-2-[l-((R)-2-methoxycarbonylamino-2-phenyl- acetyl)-pyrrolidin-2-yl]-lH-imidazol-4-yl}-phenyl)-thieno[3,2-b]thiophen-2-yl)-lH- benzoimidazol-2-yl} -pyrrolidine- l-carbonyl)-2-methyl-propyl]-carbamic acid methyl ester
[(S)-1-((S)-2-{6-[6-(4-{(S)-2-[1-((R)-2-methoxycarbonylamino-2-phenyl-acetyl)-pyrrolidin-2-yl]-3H-imidazol-4-yl}-phenyl)-thieno[3,2-b]thiophen-3-yl]-1H-benzoimidazol-2-yl}-pyrrolidine-1-carbonyl)-2-methyl-propyl]-carbamic acid methyl ester
Carbamic acid, N-((1R)-2-((2S)-2-(5-(4-(6-(2-((2S)-1-((2S)-2-((methoxycarbonyl)amino)-3-methyl-1-oxobutyl)-2-pyrrolidinyl)-1H-benzimidazol-6-yl)thieno(3,2-b)thien-3-yl)phenyl)-1H-imidazol-2-yl)-1-pyrrolidinyl)-2-oxo-1-phenylethyl)-, methyl ester
Methyl N-((1R)-2-((2S)-2-(5-(4-(6-(2-((2S)-1-((2S)-2-((methoxycarbonyl)amino)-3-methylbutanoyl)pyrrolidin-2-yl)-3H-benzimidazol-5-yl)thieno(3,2-b)thiophen-3-yl)phenyl)-1H-imidazol-2-yl)pyrrolidin-1-yl)-2-oxo-1-phenylethyl)carbamate
- Idenix Pharmaceuticals, Inc. phase 2
- CAS Number: 1312547-19-5
- C47-H48-N8-O6-S2
- 885.0782
- UNII-21P699C5FC
- deleted CAS Registry Numbers: 1458063-70-1
- INhttp://www.google.com/patents/WO2011075615A1?cl=en
A 215
compd in
http://www.google.com/patents/WO2014036244A1?cl=en
ANY ERROR amcrasto@gmail.com
samatasvir
Samatasvir is an orally-available pan-genotypic hepatitis C virus (HCV) non-structural protein 5A (NS5A) inhibitor in phase II clinical studies at Idenix for the treatment of treatment-naïve genotype 1-4 HCV-infected patients in combination with simeprevir and ribavirin.Jun 6, 2013Idenix Pharmaceuticals Announces Samatasvir (IDX719) Poster Presentations at the Asian Pacific Association for the Study of the Liver (APASL) Conference
CAMBRIDGE, Mass., June 6, 2013 (GLOBE NEWSWIRE) -- Idenix Pharmaceuticals, Inc. (Nasdaq:IDIX), a biopharmaceutical company engaged in the discovery and development of drugs for the treatment of human viral diseases, today announced three poster presentations featuring clinical and preclinical data for samatasvir (IDX719), Idenix's once-daily pan-genotypic NS5A inhibitor for the treatment of hepatitis C virus (HCV) infection, at the Asian Pacific Association for the Study of the Liver (APASL) Liver Week 2013, taking place in Singapore, June 6-10, 2013. Idenix recently initiated a phase II clinical trial (HELIX-1) evaluating an all-oral, direct-acting antiviral (DAA) HCV combination regimen of samatasvir and simeprevir (TMC435), a once-daily protease inhibitor jointly developed by Janssen R&D Ireland and Medivir AB.
The following abstracts will be presented in poster sessions during APASL Liver Week 2013 in the Conference Exhibition Hall on Friday, June 7, 2013, 8:30am - 5:30pm SGT:
- Abstract No. 2110: "Pharmacokinetics and Pharmacodynamics of IDX719, a Pan-Genotypic HCV NS5A Inhibitor, in Genotype 1, 2, 3 or 4 HCV-Infected Subjects."
- Abstract No. 2121: "Hepatitis C Virus NS5A Inhibitor IDX719 Demonstrates Potent, Pan-genotypic Activity in Preclinical and Clinical Studies."
- Abstract No. 2127: "IDX719, a Pan-genotypic HCV NS5A Replication Complex Inhibitor, Is a Promising Candidate for HCV Combination DAA Treatment."
Samatasvir is an NS5A inhibitor with low picomolar, pan-genotypic antiviral activity in vitro. To date, samatasvir has been safe and well-tolerated after single and multiple doses of up to 150 mg in healthy volunteers for up to 14 days' duration and up to 100 mg in HCV-infected patients up to 3 days' duration. There have been no treatment-emergent serious adverse events reported in the program. Samatasvir has demonstrated potent pan-genotypic antiviral activity in HCV-infected patients with mean maximal viral load reductions up to approximately 4.0 log10 IU/mL across HCV genotypes 1-4 in a proof-of-concept, three-day monotherapy study.
The HELIX-1 trial is a 12-week, randomized, double-blind, parallel group study evaluating the safety and tolerability of samatasvir and simeprevir in addition to antiviral activity endpoints, with a target enrollment of 90 treatment-naïve, non-cirrhotic, genotype 1b or 4 HCV-infected patients. The HELIX-1 trial is the first study in HCV-infected patients to commence under a non-exclusive collaboration agreement signed with Janssen in January 2013. A second trial (HELIX-2) of samatasvir, simeprevir and TMC647055, a once-daily non-nucleoside polymerase inhibitor boosted with low-dose ritonavir being developed by Janssen, is expected to initiate in the second half of 2013.
ABOUT HEPATITIS C
Hepatitis C virus is a common blood-borne pathogen infecting three to four million people worldwide annually. The World Health Organization (WHO) estimates that more than 170 million people worldwide are chronically infected with HCV, representing a nearly 5-fold greater prevalence than human immunodeficiency virus.
ABOUT IDENIX
Idenix Pharmaceuticals, Inc., headquartered in Cambridge, Massachusetts, is a biopharmaceutical company engaged in the discovery and development of drugs for the treatment of human viral diseases. Idenix's current focus is on the treatment of patients with hepatitis C virus (HCV) infection. For further information about Idenix, please refer to www.idenix.com. - .............................................
- WO 2014036244
- http://www.google.com/patents/WO2014036244A1?cl=en
- [(5)-l-((5)-2-{6-[6-(4-{(5)-2-[l-((i?)-2- methoxycarbonylamino-2-phenyl-acetyl)-pyrrolidin-2-yl]-3H-imidazol-4-yl}-phenyl)- thieno[3,2-¾]thiophen-3-yl]- lH-benzoimidazol-2-yl} -pyrrolidine- 1 -carbonyl)-2-methyl- propyl]-carbamic acid methyl ester ("the Compound"), having the structure of Formula I:(I) or an isotopic variant thereof, or a pharmaceutically acceptable salt or solvate thereof.The Compound is a nonstructural protein 5A (NS5A) inhibitor. See U.S. Pat.App. Pub. Nos. US 2011/0150827 and US 2012/0252721, the disclosure of each of which is incorporated herein by reference in its entirety. The Compound is a potent and pan-genotypic inhibitor of HCV replication in vitro, with EC50 values ranging from 2 to 24 pM against HCV genotypes la, lb, 2a, 3a, 4a, and 5a. Id.The Compound can be prepared according to the methods described in U.S.Pat. App. Pub. No. US 2011/0150827. The Compound can be also synthesized according to other methods apparent to those of skill in the art based upon the teaching herein.
- ..................
- WO 2011075615
- http://www.google.com/patents/WO2011075615A1?cl=en
- Example 36Synthesis of [(5)-l-((5)-2- {6-[5-(4- {(5)-2-[l-((R)-2-methoxycarbonylamino-2-phenyl- acetyl)-pyrrolidin-2-yl]-lH-imidazol-4-yl}-phenyl)-thieno[3,2-b]thiophen-2-yl)-lH- benzoimidazol-2-yl} -pyrrolidine- l-carbonyl)-2-methyl-propyl]-carbamic acid methyl esterA215A215
1] Compound A215 was synthesized as shown
Scheme 27
[00612] Preparation of (S 2-[6-(5-bromo-thieno[3,2,b]thiophen-2-yl)-lH- benzoimidazol-2-yl] -pyrrolidine- 1-carboxylic acid tert-butyl ester E64. In a round bottom flask were added intermediate 66 (2.42 mmol) and 3,6-dibromo-thieno[3,2-b]thiophene (7.26 mmol). The system was purged and anhydrous dioxane (36 mL) was added. Then, NaHC(¾ 1M (7.26 mmol) and Pdl 18 (0.242 mmol) were added. The reaction mixture was stirred under reflux (110 °C) for 1.5 hrs. The reaction mixture was cooled down to room temperature and DCM was added. The mixture was washed with water and the organic layer dried, filtered, and concentrated under reduced pressure. The residue was purified by silica gel chromatography (eluent: DCM to DCM/MeOH 2%) to give intermediate E64 as a yellow foam in 19% yield. MS (ESI, EI+) m/z = 505.8 (MH+).[00613] Preparation of 6-(5-bromo-thieno[3,2,b]thiophen-2-yl)-(S -2-pyrrolidin-2-yl- lH-benzoimidazole, hydrochloride E65. Intermediate E65 was synthesized from
intermediate E64 (0.198 mmol), following the procedure as described for intermediate E47 (without purification) to give intermediate E65 as a yellow solid in quantitative yield. MS (ESI, EI+) m/z = 405.8 (MH+).
[00614] Preparation of ((5)-l- {(5)-2-[6-(5-bromo-thieno[3,2-b]thiophen-2-yl)-lH- benzoimidazol-2-yl]-pyrrolidine-l-carbonyl}-2-methyl-propyl)-carbamic acid methyl ester E66. Intermediate E65 (0.198 mmol) was dissolved in anhydrous DCM (5 mL). The intermediate 1 (0.198 mmol) was added, followed by HATU (0.257 mmol) and Et3N (0.792 mmol). The reaction mixture was stirred at room temperature for 45 min. DCM was added and the mixture was washed with water. The organic layer was dried over a2S04, filtered, and concentrated under reduced pressure. The residue was purified by silica gel
chromatography (eluent: DCM to DCM/MeOH 2%) to give intermediate E66 in quantitative yield. MS (ESI, EI+) m/z = 562.7 (MH+).
[00615] Preparation of (S 2-{4-[4-(5-{(5')-2-[l-((5,)-2-methoxycarbonylamino-3- methyl-butyryl)-pyrrolidin-2-yl]-3H-benzoimidazol-5-yl}-thieno[3,2-b]thiophen-2-yl)- phenyl]-lH-imidazol-2-yl}-pyrrolidine-l-carboxylic acid tert-butyl ester E67. Intermediate E67 was synthesized from intermediate E66 (0.196 mmol), following the procedure as described for the compound Al (1 10° C for 35 min). The residue was purified by silica gel chromatography (eluent: DCM to DCM/MeOH 4%) to give intermediate E67 as a yellow solid in 46% yield. MS (ESI, EI ) m/z = 794.2 (MH ).
[00616] Preparation of{2-methyl-(5)-l-[(5)-2-(6-{5-[4-((5)-2-pyrrolidin-2-yl-lH- imidazol-4-yl)-phenyl]-thieno[3,2-b]thiophen-2-yl}-lH-benzoimidazol-2-yl)-pyrrolidine-l- carbonyl]-propyl}-carbamic acid methyl ester, hydrochloride E68. Intermediate E68 was synthesized from intermediate E67 (0.086 mmol), following the procedure as described for intermediate E47 (without purification) to give intermediate E68 as an orange solid in quantitative yield. MS (ESI, EI+) m/z = 694.14 (MH+).
[00617] Preparation of [(5)-l-((5)-2- {6-[5-(4- {(5)-2-[l-((R)-2-methoxycarbonylamino- 2-phenyl-acetyl)-pyrrolidin-2-yl]-lH-imidazol-4-yl}-phenyl)-thieno[3,2-b]thiophen-2-yl)- lH-benzoimidazol-2-yl} -pyrrolidine- l-carbonyl)-2-methyl-propyl]-carbamic acid methyl ester A215. Compound A215 was synthesized from intermediate E68 (0.086 mmol) following the procedure as described for compound A114 to give compound A215 as a yellow solid in 48% yield. H NMR (DMS0-< 400 MHz) δ (ppm) 0.82 (d, J= 6.70 Hz, 3H), 0.86 (d, J= 6.70 Hz, 3H), 1.82-2.10 (m, 7H), 2.16-2.28 (m, 2H), 3.10-3.16 (m, 1H), 3.52-3.55 (m, 6H), 3.80-3.90 (m, 3H), 4.07 (t, J= 8.38 Hz, 1H), 5.04-5.19 (m, 2H), 5.37-5.53 (m, 1H), 6.91-7.1 (m, 1H), 7.30-7.88 (m, 15H), 11.77-1.95 (m, 1H), 12.29 (brs, 1H); MS (ESI, EI+) m/z = 885.3 (MH+). - ................
- WO 201213558







.................
US 2013071352\
http://www.google.com/patents/US20130071352
Example 36 Synthesis of [(S)-1-((S)-2-{6-[5-(4-{(S)-2-[1-((R)-2-methoxycarbonylamino-2-phenyl-acetyl)-pyrrolidin-2-yl]-1H-imidazol-4-yl}-phenyl)-thieno[3,2-b]thiophen-2-yl)-1H-benzoimidazol-2-yl}-pyrrolidine-1-carbonyl)-2-methyl-propyl]-carbamic acid methyl ester A215

Compound A215 was synthesized as shown in 27.


Preparation of (S)-2-[6-(5-bromo-thieno[3,2,b]thiophen-2-yl)-1H-benzoimidazol-2-yl]-pyrrolidine-1-carboxylic acid tert-butyl ester E64. In a round bottom flask were added intermediate 66 (2.42 mmol) and 3,6-dibromo-thieno[3,2-b]thiophene (7.26 mmol). The system was purged and anhydrous dioxane (36 mL) was added. Then, NaHCO3 1M (7.26 mmol) and Pd118 (0.242 mmol) were added. The reaction mixture was stirred under reflux (110° C.) for 1.5 hrs. The reaction mixture was cooled down to room temperature and DCM was added. The mixture was washed with water and the organic layer dried, filtered, and concentrated under reduced pressure. The residue was purified by silica gel chromatography (eluent: DCM to DCM/MeOH 2%) to give intermediate E64 as a yellow foam in 19% yield. MS (ESI, EI+) m/z=505.8 (MH+).
Preparation of 6-(5-bromo-thieno[3,2,b]thiophen-2-yl)-(S)-2-pyrrolidin-2-yl-1H-benzoimidazole, hydrochloride E65. Intermediate E65 was synthesized from intermediate E64 (0.198 mmol), following the procedure as described for intermediate E47 (without purification) to give intermediate E65 as a yellow solid in quantitative yield. MS (ESI, EI+) m/z=405.8 (MH+).
Preparation of ((S)-1-{(S)-2-[6-(5-bromo-thieno[3,2-b]thiophen-2-yl)-1H-benzoimidazol-2-yl]-pyrrolidine-1-carbonyl}-2-methyl-propyl)-carbamic acid methyl ester E66. Intermediate E65 (0.198 mmol) was dissolved in anhydrous DCM (5 mL). The intermediate 1 (0.198 mmol) was added, followed by HATU (0.257 mmol) and Et3N (0.792 mmol). The reaction mixture was stirred at room temperature for 45 min. DCM was added and the mixture was washed with water. The organic layer was dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by silica gel chromatography (eluent: DCM to DCM/MeOH 2%) to give intermediate E66 in quantitative yield. MS (ESI, EI+) m/z=562.7 (MH+).
Preparation of (S)-2-{4-[4-(5-{(S)-2-[1-((S)-2-methoxycarbonylamino-3-methyl-butyryl)-pyrrolidin-2-yl]-3H-benzoimidazol-5-yl}-thieno[3,2-b]thiophen-2-yl)-phenyl]-1H-imidazol-2-yl}-pyrrolidine-1-carboxylic acid tert-butyl ester E67. Intermediate E67 was synthesized from intermediate E66 (0.196 mmol), following the procedure as described for the compound A1 (110° C. for 35 min). The residue was purified by silica gel chromatography (eluent: DCM to DCM/MeOH 4%) to give intermediate E67 as a yellow solid in 46% yield. MS (ESI, EI+) m/z=794.2 (MH+).
Preparation of{2-methyl-(S)-1-[(S)-2-(6-{5-[4-((S)-2-pyrrolidin-2-yl-1H-imidazol-4-yl)-phenyl]-thieno[3,2-b]thiophen-2-yl}-1H-benzoimidazol-2-yl)-pyrrolidine-1-carbonyl]-propyl}-carbamic acid methyl ester, hydrochloride E68. Intermediate E68 was synthesized from intermediate E67 (0.086 mmol), following the procedure as described for intermediate E47 (without purification) to give intermediate E68 as an orange solid in quantitative yield. MS (ESI, EI+) m/z=694.14 (MH+).
Preparation of [(S)-1-((S)-2-{6-[5-(4-{(S)-2-[1-((R)-2-methoxycarbonylamino-2-phenyl-acetyl)-pyrrolidin-2-yl]-1H-imidazol-4-yl}-phenyl)-thieno[3,2-b]thiophen-2-yl)-1H-benzoimidazol-2-yl}-pyrrolidine-1-carbonyl)-2-methyl-propyl]-carbamic acid methyl ester A215. Compound A215 was synthesized from intermediate E68 (0.086 mmol) following the procedure as described for compound A114 to give compound A215 as a yellow solid in 48% yield. 1H NMR (DMSO-d6, 400 MHz) δ (ppm) 0.82 (d, J=6.70 Hz, 3H), 0.86 (d, J=6.70 Hz, 3H), 1.82-2.10 (m, 7H), 2.16-2.28 (m, 2H), 3.10-3.16 (m, 1H), 3.52-3.55 (m, 6H), 3.80-3.90 (m, 3H), 4.07 (t, J=8.38 Hz, 1H), 5.04-5.19 (m, 2H), 5.37-5.53 (m, 1H), 6.91-7.1 (m, 1H), 7.30-7.88 (m, 15H), 11.77-1.95 (m, 1H), 12.29 (brs, 1H); MS (ESI, EI+) m/z=885.3 (MH+).
..........................
US2012/252721
Example 33Synthesis of [(S)-1-((S)-2-{6-[6-(4-{(S)-2-[1-((R)-2-methoxycarbonylamino-2-phenyl-acetyl)-pyrrolidin-2-yl]-3H-imidazol-4-yl}-phenyl)-thieno[3,2-b]thiophen-3-yl]-1H-benzoimidazol-2-yl}-pyrrolidine-1-carbonyl)-2-methyl-propyl]-carbamic acid methyl ester A169
 SEEMS LIKE AN ERROR, benzene ring
SEEMS LIKE AN ERROR, benzene ring
Compound 169 was synthesized as shown in Scheme 24.

Preparation of (S)-2-{5-[4-(6-bromo-thieno[3,2-b]thiophen-3-yl)-phenyl]-1H-imidazol-2-yl}-pyrrolidine-1-carboxylic acid tert-butyl ester E78. To a mixture of DMF and water (20 mL/2.5 mL) were added Pd(PPh3)4 (0.1 mmol), 3,6-dibromo-thieno[3,2-b]thiophene (1.01 mmol), intermediate 6 (1.1 mmol), and sodium carbonate (4.04 mmol). The reaction mixture was degassed and irradiated for 1 hr at 80° C. Ethyl acetate was added and the organic layer was washed with water. The organic layer was dried over Na2SO4, filtered, and evaporated in vacuo. The residue was purified by silica gel chromatography (eluent: DCM-DCM/MeOH 98/2) to give intermediate E78 as a green gum in 41% yield. MS (ESI, EI+) m/z=532.19-530.31 (MH+).
Preparation of (S)-2-{5-[4-(6-{(S)-2-[1-((S)-2-methoxycarbonylamino-3-methyl-butyryl)-pyrrolidin-2-yl]-3H-benzoimidazol-5-yl}-thieno[3,2-b]thiophen-3-yl)-phenyl]-1H-imidazol-2-yl}-pyrrolidine-1-carboxylic acid tert-butyl ester E79. Compound 78 (0.198 mmol), intermediate 83 (0.228 mmol), and 1,1′-bis(di-tert-BP)ferrocene palladium dichloride (0.03 mmol) were added to a solution of dioxane (4 mL) and 1M NaHCO3 in water (0.594 mmol). The reaction mixture was irradiated at 90° C. for 1 hr. The mixture was diluted in dichloromethane and washed with water. The two layers were separated and the organic layer was concentrated under reduced pressure. The residue was purified by silica gel chromatography (eluent: DCM-DCM/MeOH 95/5) to give intermediate E79 as a brown foam in 70% yield. 1H NMR (CDCl3, 400 MHz) δ (ppm) 0.90-0.91 (m, 6H), 1.51 (s, 9H), 1.67-2.40 (m, 10H), 3.07-3.1 (m, 2H), 3.45-3.50 (m, 1H), 3.72 (s, 3H), 3.90 (m, 1H), 4.37 (m, 1H), 5.00-5.01 (m, 1H), 5.45-5.48 (m, 2H), 7.26-8.12 (m, 10H), 10.67 (m, 1H); MS (ESI, EI+) m/z=792.79 (MH−).
Preparation of [(S)-1-((S)-2-{6-[6-(4-{(S)-2-[1-((R)-2-methoxycarbonylamino-2-phenyl-acetyl)-pyrrolidin-2-yl]-3H-imidazol-4-yl}-phenyl)-thieno[3,2-b]thiophen-3-yl]-1H-benzoimidazol-2-yl}-pyrrolidine-1-carbonyl)-2-methyl-propyl]-carbamic acid methyl ester A169. Intermediate E79 (0.132 mmol) was dissolved in methanol (2.6 mL) and 4N HCl in dioxane (2.64 mL) was added. The mixture was stirred 1 hr at room temperature before concentration under reduced pressure. The residue was dissolved in DMF (2.6 mL) and the mixture was cooled down to −100C. TEA (0.924 mmol), intermediate 31 (0.139 mmol), and HATU (0.172 mmol) were added and the mixture was stirred at −100C for 1 hr. Ethyl acetate was added and the mixture was washed with water. The organic layer was dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was filtered on a SCX-2 column and purified by silica gel chromatography (eluent: DCM-DCM/MeOH 97/3) to give compound A169 as a beige solid in 74% yield.
1H NMR (CDCl3, 400 MHz) δ (ppm) 0.89-0.91 (m, 6H), 1.40-2.42 (m, 8H), 3.08-3.24 (m, 3H), 3.67 (m, 3H), 3.71 (m, 4H), 3.88-3.89 (m, 1H), 4.34-4.38 (m, 1H), 5.30-5.32 (m, 1H), 5.42-5.45 (m, 3H), 6.03-6.04 (m, 1H), 7.26-8.14 (m, 16H), 10.65 (m, 1H);
MS (ESI, EI+) m/z=885.8 (MH+).
| US20110150827 * | Dec 17, 2010 | Jun 23, 2011 | Idenix Pharmaceuticals, Inc. | 5,5-fused arylene or heteroarylene hepatitis c virus inhibitors |
| US20120252721 * | Mar 29, 2012 | Oct 4, 2012 | Idenix Pharmaceuticals, Inc. | Methods for treating drug-resistant hepatitis c virus infection with a 5,5-fused arylene or heteroarylene hepatitis c virus inhibitor |
24
PALINAVIR












 8 was added to NaOH and alkylated with 4-picolyl chloride. The protecting group was lost with the addition of acid.
8 was added to NaOH and alkylated with 4-picolyl chloride. The protecting group was lost with the addition of acid.
 Derivation of 9 was started by a simple substitution of 19, quinoline-2-carboxylic acid, to 20, an acid chloride, with the help of thionyl chloride. Acylation of amino acid L-valine to 20 was accomplished by a biphasic system.
Derivation of 9 was started by a simple substitution of 19, quinoline-2-carboxylic acid, to 20, an acid chloride, with the help of thionyl chloride. Acylation of amino acid L-valine to 20 was accomplished by a biphasic system.
 In the original synthesis of palinavir, a 2:1 mixture of 3 to 5 was needed to produce only ~35% of 6 and flash chromatography was needed. On a large scale without chromatography, 6 was produced with a 85% yield, but 21 was also produced. To keep the production of 21 to a minimum, the reaction was performed in a solution that was degassed. This insured that the pyridine ring would not react in the presence of air. With this precaution, only 1-2% of the yield was 21. A washing of the solution with 1 M KH2PO4 removed and left over 5. Deprotection was achieved with the addition of concentrated HCl and followed by adding NaOH. The product of 10 was a “viscous syrup”. 22 was 1-1.5% of the product and was not removed before the addition of 9 to form 80-85% palinavir.
In the original synthesis of palinavir, a 2:1 mixture of 3 to 5 was needed to produce only ~35% of 6 and flash chromatography was needed. On a large scale without chromatography, 6 was produced with a 85% yield, but 21 was also produced. To keep the production of 21 to a minimum, the reaction was performed in a solution that was degassed. This insured that the pyridine ring would not react in the presence of air. With this precaution, only 1-2% of the yield was 21. A washing of the solution with 1 M KH2PO4 removed and left over 5. Deprotection was achieved with the addition of concentrated HCl and followed by adding NaOH. The product of 10 was a “viscous syrup”. 22 was 1-1.5% of the product and was not removed before the addition of 9 to form 80-85% palinavir.




PALINAVIR
PALINAVIR, BILA-2011-BS
UNII-632S1WU9Z2, 154612-39-2, n-[(1s)-1-[[(1s,2r)-1-benzyl-3-[(2s,4r)-2-(tert-butylcarbamoyl)-4-(4-pyridylmethoxy)piperidino]-2-hydroxypropyl]carbamoyl]-2-methylpropyl]quinaldamide,
N-[(2S)-1-[[(2S,3R)-4-[(2S,4R)-2-(tert-butylcarbamoyl)-4-(pyridin-4-ylmethoxy)piperidin-1-yl]-3-hydroxy-1-phenylbutan-2-yl]amino]-3-methyl-1-oxobutan-2-yl]quinoline-2-carboxamide
Molecular Formula:C41H52N6O5
Molecular Weight:708.88878 g/mol
| PATENT | SUBMITTED | GRANTED |
|---|---|---|
| Substituted pipecolinic acid derivatives as HIV protease inhibitors [US5614533] | 1997-03-25 | |
| Substituted pipecolinic acid derivatives as HIV protease inhibitors. [EP0560268] | 1993-09-15 | 1995-01-04 |
……………………….
PATENT
Scheme 5: Synthesis of Palinavir (6):

The organic solvent mentioned according to the invention is selected from the group consisting of organic solvents, wherein the organic solvents are polar aprotic such as DCM, THF, Ethyl acetate, acetone, DMF, acetonitrile, DMSO ; polar protic solvents such as lower alcohol particularly (C1-C6) alkyl alcohol, water, acetic acid ; non-polar solvents such as hexane, benzene, toluene, chloroform, pet. ether, 1,4-dioxane, heptane either alone or mixtures thereof . Additionally the purification or separation of crude product can be accomplished by known techniques viz. extraction, column chromatography in a suitable organic solvent with the aid of instruments such as TLC, HPLC, GC, mass spectroscopy, or distillation, crystallization, derivatization.
………………………….
J Org Chem 1997,62(11),3440
The reaction of tert-butoxycarbonyl-L-phenylalanine (I) with isobutyl chloroformate in THF gives the expected mixed anhydride which is treated with diazomethane and HCl yielding the corresponding chloromethyl ketone (II). The reduction of (II) with NaBH4 in THF affords the (S)-chlorohydrin (IV), which is treated with KOH in ethanol to obtain the chiral epoxide (V)(1,2). Ring opening of (V) with (?(cis)-N-tert-butyl-4-(4-pyridylmethoxy)piperidine-2-carboxamide (VI) by a treatment with LiCl in refluxing ethanol gives a mixture of diastereomers that is separated by chromatography giving the pure isomer (VII). The reaction of (VII) with tert-butoxycarbonyl-L-valine (VIII) by treatment first with trifluoroacetic acid (TFA), and condesation by means of BOP ((benzotriazol-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate) and NMM (N-methylmorpholine) affords the expected condensation product (IX). Finally, this compound is condensed with quinoline-2-carboxylic acid (X) by means of BOP and NMM as before. 2) The piperidine (VI) has been obtained by condensation of (?(cis)-N-(tert-butoxycarbonyl)-4-hydroxypiperidine-2-carboxamide (XI) with 4-(chloromethyl)pyridine (XII) by means of NaH in DMS, followed by hydrolysis with HCl.
Palinavir can also be obtained as follows: The controlled oxidation of 2(S)-(dibenzylamino)-3-phenyl-1-propanol (XIII) with pyridine-SO3 complex in DMSO gives the corresponding aldehyde (XIV), which is condensed with bromochloromethane (XV) by means of Li in THF followed by hydrolysis with HCl yielding regioselectively the 1-chloro-2-butanol (XVI). The debenzylation of (XVI) by hydrogenation over Pd/C affords the free amine (XVII), which is treated with tert-butoxycarbonyl anhydride/triethylamine and dehydrochlorinated with KOH in methanol to give the desired chiral epoxide (V).
The chiral piperidine (2S,4R)(VI) has been obtained as follows: The cyclization of 3-buten-1-ol (XXII) with (S)-1-phenylethylamine (XXIII) and glyoxylic acid (XXIV) by means of tosyl chloride in THF gives a mixture of the (2S,4R) and (2R,4S) lactones (XXV), which is resolved by fractional crystallyzation of their salts with the chiral camphorsulfonic acid (XXVI), followed by elimination of the acid with ammonia to afford (2S,4R)(XXVII). The reaction of lactone (XXVII) with isopropylmagnesium chloride and tert-butylamine in THF gives (2S,4R)-N-tert-butyl-4-hydroxy-1-(1(S)-phenylethyl)piperidine-2-carboxamide (XXVIII), which is debenzylated by hydrogenation and protected with tert-butoxycarbonyl anhydride yielding (2S,4R)-N-(tert-butoxycarbonyl)-4-hydroxypiperidine-2-carboxamide (2S,4R)(XI), which is finally condensed with 4-(chloromethyl)pyridine (XII) as before to obtain the chiral piperidine (2S,4R)(VI), already reported.
The condendsation of epoxide (V) with (2S,4R)(VI) by means of basic alumina in THF, followed by elimination of the protecting group with HCl and NaOH yields directly the condensation product (XVIII) as a pure diastereomer and with a free amino group. Finally, this compound is condensed with N-(2-quinolylcarbonyl)-L-valine (XIX) through its activation compound with isobutyl chloroformate (the 4(S)-isopropyl-2-(2-quinolyl)oxazol-5(4H)-one (XX)). The N-acyl-L-valine (XIX) has been obtained by acylation of L-valine (XXI) with quinoline-2-carboxylic acid (X) through its acyl chloride obtained with SOCl2.
………………………..
Palinavir is an inhibitor with five chiral centers. It contains the amino acid valine and pipecolinin acid. The previous way to create this drug faced three major obstacles. First, the reaction from 2 to 3 used diazomethane. Therefore, is is difficult, if not impossible, to produce large quantities. Secondly, the steps included in going from 4 to 5 gave way to racemers which is very inefficient. Finally, chromatography is needed at two separate times.
Four issues were addresses in route to product 1. First, because of the number of chiral centers, stereochemical control was a concern. high chemical yields were a second concern. Also, multi step procedures were advantageous to cut down on purification steps. Finally, the synthesis tried to restrict the use of hazardous reagents. The following retrosynthesis reaction was conceived and three target molecules were identified as seen in figure 1.
Molecule 3 uses a diaseteroselective addition of in situ (chloromethyl)lithium to N,N-dibenzylphenylalaninol and is derived from a four step process.
Recrystallization of 13 is required. Molecule 14 was not reached because it posed a problem later in the reaction. The N-benzyl protection group could not be removed to react with 9.
8 is a derivative of naturally occurring pipocolic acid, 16, named 3-buten-1-ol. Selective crystallization of diastereomeric salts can lead to 17a, but a more efficient way is by having a 60:40 mixture of lactones 17a,b. This leads to 18a,b using a Brodroux process. Crystallization of 18a,b lead to a poor overall yield. Instead, 18a,b undergoes salt crystallization with (-)-camphorsulfonic acid. Finally, 18a underwent hydrolysis and then addition of di-tert butyl dicarbonate leads to 8.
8 was then transformed to 5 in a three step process.
8 was added to NaOH and alkylated with 4-picolyl chloride. The protecting group was lost with the addition of acid. Derivation of 9 was started by a simple substitution of 19, quinoline-2-carboxylic acid, to 20, an acid chloride, with the help of thionyl chloride. Acylation of amino acid L-valine to 20 was accomplished by a biphasic system.In the original synthesis of palinavir, a 2:1 mixture of 3 to 5 was needed to produce only ~35% of 6 and flash chromatography was needed. On a large scale without chromatography, 6 was produced with a 85% yield, but 21 was also produced. To keep the production of 21 to a minimum, the reaction was performed in a solution that was degassed. This insured that the pyridine ring would not react in the presence of air. With this precaution, only 1-2% of the yield was 21. A washing of the solution with 1 M KH2PO4 removed and left over 5. Deprotection was achieved with the addition of concentrated HCl and followed by adding NaOH. The product of 10 was a “viscous syrup”. 22 was 1-1.5% of the product and was not removed before the addition of 9 to form 80-85% palinavir.
Coupling of 10 and 9 is the final step in the synthesis , although there are still some purification steps left.
Two recrystallizations were required for the final 99.6% purity.
………………………..
J. Org. Chem., 1997, 62 (11), pp 3440–3448
DOI: 10.1021/jo9702655
Palinavir is a potent peptidomimetic-based HIV protease inhibitor. We have developed a highly convergent and stereoselective synthesis which is amenable to the preparation of multikilogram quantities of this compound. The synthetic sequence proceeds in 24 distinct chemical steps (with several integrated, multistep operations) from commercially available starting materials. No chromatographies are required throughout the process, and the final product is purified by crystallization of its dihydrochloride salt to >99% homogeneity.
crude palinavir (1) as a thick brown oil (yield not determined). HPLC analysis (Supelcosil LZ-ABZ, 10−50% 1% TFA in MeCN/1% TFA in 25 min, 1 mL/min flow rate): 1, tR 17.80 min (84.1%); 24, tR 18.47 min (2.0%); 25, tR 19.97 min (1.45%).
palinavir dihydrochloride (1750 g, 51% yield) containing 0.25% w/w isopropanol (by 1H NMR):
mp 175−185 °C.
[α]25D −13.0° (c 1, MeOH). [α]25Hg365 +44.9° (c 1, MeOH).
IR (KBr) ν 3700−2300, 1660, 1555, 1520 cm-1.
1H NMR (DMSO-d6) δ 10.00 (broad s, 1H), 8.88 (d, J = 6.3 Hz, 2H), 8.61 (d, J= 8.4 Hz, 1H), 8.60 (s, 1H), 8.51 (d, J = 9.6 Hz, 1H), 8.35 (d, J = 8.7 Hz, 1H), 8.20 (d, J = 8.4 Hz, 1H), 8.16 (d, J = 8.7 Hz, 1H), 8.11 (d, J = 8.1 Hz, 1H), 7.94 (d, J = 6.0 Hz, 2H), 7.89 (t, J = 7.6 Hz, 1H), 7.74 (t, J = 7.5 Hz, 1H), 7.19 (d, J = 7.2 Hz, 2H), 7.08 (t, J = 7.5 Hz, 2H), 6.91 (t, J = 7.3 Hz, 1H), 4.86 (AB quartet, 2H), 4.37 (broad t, J = 7.8 Hz, 1H), 4.21 (d, J = 11.4 Hz, 1H), 4.11 (broad m, 1H), 3.96 (broad m, 1H), 3.80−3.65 (m, 2H), 3.26 (t, J = 7.4 Hz, 1H), 3.15−3.01 (m, 2H), 2.94 (broad d, J = 12.0 Hz, 1H), 2.62 (dd, J = 13.6, 10.6 Hz, 1H), 2.56 ((broad d, J = 12.0 Hz, 1H), 2.20−2.05 (m, 2H), 1.86 (m, 1H), 1.69 (q, J = 11.7 Hz, 1H), 1.31 (s, 9H), 0.81 (d, J = 6.3 Hz, 3H), 0.80 (d, J = 6.6 Hz, 3H).
13C NMR (DMSO-d6) δ 170.4, 166.4, 163.3, 158.3, 149.5, 145.9, 141.9, 138.6, 138.2, 130.7, 129.3, 129.1, 129.0, 128.3, 128.2, 128.0, 125.9, 124.1, 118.6, 72.3, 68.8, 67.2, 64.8, 58.0, 57.8, 54.4, 51.3, 51.1, 35.4, 34.1, 31.1, 28.2, 19.5, 17.9.
FAB-MS m/z 709 (MH+ of free base). Anal. Calcd for C41H54Cl2N6O5 (corrected for 8% water content as determined by Karl Fisher analysis and 0.25% w/w isopropanol as determined by 1H NMR): C, 58.31; H, 7.29; N, 9.93. Found: C, 57.76; H, 7.25; N, 9.89. Titration of HCl content using NaOH: 2.09 ± 0.03 mol HCl. HPLC homogeneity (Supelcosil LC-ABZ, 10−50% 1% TFA in MeCN/1% TFA in 25 min, 1 mL/min flow rate): palinavir dihydrochloride, tR 18.24 min (99.51%); 25 tR 20.39 min (0.33%). HPLC homogeneity (Nova-Pak C8, 20−80% MeCN/50 mM NaH2PO4 in 25 min, 1 mL/min flow rate): palinavir dihydrochloride, tR 15.52 min (99.67%); 25 tR 13.52 min (0.33%).
PURE palinavir (1) as a white amorphous powder (1902 g, 84% yield):
mp 100−107 °C. [α]25D −11.5° (c 1, MeOH).
IR (KBr) ν 3700−3100, 1660, 1520, 1495 cm-1.
1H NMR (CDCl3) δ 8.54 (d, J = 5.7 Hz, 2H), 8.48 (d, J = 8.6 Hz, 1H), 8.31 (d, J= 8.6 Hz, 1H, part of AB), 8.22 (d, J = 8.3 Hz, 1H, part of AB), 8.13 (d, J = 8.3 Hz, 1H), 7.90 (d, J = 8.0 Hz, 1H), 7.80 (t, J = 7.6 Hz, 1H), 7.65 (t, J = 7.6 Hz, 1H), 7.25 (d, J = 5.4 Hz, 2H), 7.13 (d, J = 7.3 Hz, 2H), 7.07 (t, J = 7.5 Hz, 1H), 6.92 (t, J = 7.3 Hz, 1H), 6.59 (d, J = 8.3 Hz, 1H), 6.57 (s, 1H), 4.61 (d, J = 13.4 Hz, 1H, part of AB), 4.51 (d, J = 13.4 Hz, 1H, part of AB), 4.32 (dd, J = 8.6, 6.4 Hz, 1H), 4.22 (m, 1H), 3.97 (m, 1), 3.47−3.33 (m, 2H), 2.94 (dd, J = 14.3, 4.1 Hz, 1H), 2.89 (d, J= 8.6 Hz, 1H), 2.79−2.72 (m, 1H), 2.77 (dd, J = 14.3, 10.8 Hz, 1H), 2.43 (dd, J = 13.4, 8.3 Hz, 1H), 2.40−2.25 (m, 3H), 1.95 (broad d, J = 12.4 Hz, 1H), 1.65 (q J = 11.8 Hz, 2H), 1.32 (s, 9H), 0.95 (d, J = 7.0 Hz, 3H), 0.83 (d, J = 6.7 Hz, 3H).
13C NMR (CDCl3) δ 171.6, 171.2, 165.0, 149.8, 148.8, 147.9, 146.5, 137.6, 137.5, 130.3, 129.9, 129.5, 129.4, 129.0, 128.8, 128.5, 128.2, 127.7, 126.4, 121.7, 118.8, 75.0, 71.9, 68.1, 66.7, 59.4, 56.9, 54.6, 50.9, 50.2, 34.8, 33.3, 29.8, 29.7, 28.7, 19.6, 17.5.
FAB-MS m/z 709 (MH+). Anal. Calcd for C41H52N6O5(corrected for 0.7% water content as determined by Karl Fisher analysis): C, 68.98; H, 7.42; N, 11.77. Found: C, 68.71; H, 7.47; N, 11.71. HPLC homogeneity (Supelcosil LC-ABZ, 10−50% 1% TFA in MeCN/1% TFA in 25 min, 1 mL/min flow rate): palinavir (1), tR 17.83 min (99.59%); 25 tR20.00 min (0.41%). HPLC homogeneity (Nova-Pak C8, 10−80% MeCN/50 mM NaH2PO4 in 25 min, 1 mL/min flow rate): palinavir (1), tR 17.37 min (99.51%); 25 tR 15.87 min (0.49%).
| REFERENCE | ||
|---|---|---|
| 1 | * | ARUN K. GHOSH ET AL: “The Development of Cyclic Sulfolanes as Novel and High-Affinity P2 Ligands for HIV-1 Protease Inhibitors“, JOURNAL OF MEDICINAL CHEMISTRY, vol. 37, no. 8, 1 April 1994 (1994-04-01), pages 1177-1188, XP055057710, ISSN: 0022-2623, DOI: 10.1021/jm00034a016 |
| 2 | * | KAY BRICKMANN ET AL: “Synthesis of Conformationally Restricted Mimetics of [gamma]-Turns and Incorporation into Desmopressin, an Analogue of the Peptide Hormone Vasopressin“, CHEMISTRY – A EUROPEAN JOURNAL, vol. 5, no. 8, 2 August 1999 (1999-08-02), pages 2241-2253, XP055057517, ISSN: 0947-6539, DOI: 10.1002/(SICI)1521-3765(19990802)5:8<2241: :AID-CHEM2241>3.0.CO;2-L |
| 3 | * | KIRAN I N C ET AL: “A concise enantioselective synthesis of (+)-goniodiol and (+)-8-methoxygoniodiol via Co-catalyzed HKR of anti-(2SR, 3RS)-3-methoxy-3-phenyl-1, 2-epoxypropane“, TETRAHEDRON LETTERS, ELSEVIER, AMSTERDAM, NL, vol. 52, no. 3, 19 January 2011 (2011-01-19), pages 438-440, XP027558447, ISSN: 0040-4039 [retrieved on 2010-12-14] |
| 4 | * | M. TOKUNAGA: “Asymmetric Catalysis with Water: Efficient Kinetic Resolution of Terminal Epoxides by Means of Catalytic Hydrolysis“, SCIENCE, vol. 277, no. 5328, 15 August 1997 (1997-08-15), pages 936-938, XP055057541, ISSN: 0036-8075, DOI: 10.1126/science.277.5328.936 |
| 5 | * | PARKES K E B ET AL: “STUDIES TOWARD THE LARGE-SCALE SYNTHESIS OF THE HIV PROTEINASE INHIBITOR RO 31-8959“, JOURNAL OF ORGANIC CHEMISTRY, ACS, US, vol. 59, no. 13/16, 1 January 1994 (1994-01-01), pages 3656-3664, XP002011975, ISSN: 0022-3263, DOI: 10.1021/JO00092A026 |
| 6 | * | R. SANTHOSH REDDY ET AL: “Co(iii)(salen)-catalyzed HKR of two stereocentered alkoxy- and azido epoxides: a concise enantioselective synthesis of (S,S)-reboxetine and (+)-epi-cytoxazone“, CHEMICAL COMMUNICATIONS, vol. 46, no. 27, 1 January 2010 (2010-01-01), page 5012, XP055057537, ISSN: 1359-7345, DOI: 10.1039/c0cc00650e |
| 7 | * | SHINJI NAGUMO ET AL: “Intramolecular Friedel-Crafts type reaction of vinyloxiranes linked to an ester group“, TETRAHEDRON, vol. 65, no. 47, 1 November 2009 (2009-11-01), pages 9884-9896, XP055057655, ISSN: 0040-4020, DOI: 10.1016/j.tet.2009.09.037 |
| 8 | * | SUNITA K. GADAKH ET AL: “Enantioselective synthesis of HIV protease inhibitor amprenavir via Co-catalyzed HKR of 2-(1-azido-2-phenylethyl)oxirane“, TETRAHEDRON: ASYMMETRY, vol. 23, no. 11-12, 1 June 2012 (2012-06-01), pages 898-903, XP055057475, ISSN: 0957-4166, DOI: 10.1016/j.tetasy.2012.06.003 |
Amprenavir
KVX-478, 141W94, VX-478,
(3S)-Tetrahydro-3-furanyl ((1S,2R)-3-(((4-aminophenyl)sulfonyl)(2-methylpropyl)amino)-2-hydroxy-1-(phenylmethyl)propyl)carbamate
(3S)-tetrahydro-3-furyl N-[(1S,2R)-3-(4-amino-N-isobutylbenzenesulphonamido)-1-benzyl-2-hydroxypropyl] carbamate
CAS NO. 161814-49-9, [(3S)-oxolan-3-yl] N-[(2S,3R)-4-[(4-aminophenyl)sulfonyl-(2-methylpropyl)amino]-3-hydroxy-1-phenylbutan-2-yl]carbamate
| 161814-49-9 | |
| WEIGHT | 505.224656557 |
|---|---|
| CHEMICAL FORMULA | C25H35N3O6S |
| Amprenavir is a protease inhibitor used to treat HIV infection. |
Amprenavir (Agenerase, GlaxoSmithKline) is a protease inhibitor used to treat HIV infection. It was approved by the Food and Drug Administration on April 15, 1999, for twice-a-day dosing instead of needing to be taken every eight hours. The convenient dosing came at a price, as the dose required is 1,200 mg, delivered in eight very large gel capsules.
Production of amprenavir was discontinued by the manufacturer December 31, 2004; a prodrug version (fosamprenavir) is available.
| Amprenavir is a protease inhibitor with activity against Human Immunodeficiency Virus Type 1 (HIV-1). Protease inhibitors block the part of HIV called protease. HIV-1 protease is an enzyme required for the proteolytic cleavage of the viral polyprotein precursors into the individual functional proteins found in infectious HIV-1. Amprenavir binds to the protease active site and inhibits the activity of the enzyme. This inhibition prevents cleavage of the viral polyproteins resulting in the formation of immature non-infectious viral particles. Protease inhibitors are almost always used in combination with at least two other anti-HIV drugs. |

| COUNTRY | PATENT NUMBER | APPROVED | EXPIRES (ESTIMATED) |
|---|---|---|---|
| United States | 5585397 | 1993-12-17 | 2013-12-17 |
| United States | 6730679 | 1997-11-11 | 2017-11-11 |
Background
Research aimed at development of renin inhibitors as potentialantihypertensive agents had led to the discovery of compounds that blocked the action of this peptide cleaving enzyme. The amino acid sequence cleaved by renin was found to be fortuitously the same as that required to produce the HIV peptide coat. Structure–activity studies on renin inhibitors proved to be of great value for developing HIV protease inhibitors. Incorporation of an amino alcohol moiety proved crucial to inhibitory activity for many of these agents. This unit is closely related to the one found in the statine, an unusual amino acid that forms part of the pepstatin, a fermentation product that inhibits protease enzymes.
Synthesis
R.D. Tung, M.A. Murcko, G.R. Bhisetti, U.S. Patent 5,558,397 (1996). The scheme shown here is partly based on that used to prepare darunavir andfosamprenavir due to difficulty in deciphering the patent.
AGENERASE (amprenavir) is an inhibitor of the human immunodeficiency virus(HIV) protease. The chemical name of amprenavir is (3S)-tetrahydro-3-furyl N-[(1S,2R)-3-(4-amino-N-isobutylbenzenesulfonamido)-1-benzyl-2-hydroxypropyl]carbamate. Amprenavir is a single stereoisomer with the (3S)(1S,2R) configuration. It has a molecular formula of C25H35N3O6S and a molecular weight of 505.64. It has the following structural formula:
 |
Amprenavir is a white to cream-colored solid with a solubility of approximately 0.04 mg/mL in water at 25°C.
AGENERASE Capsules (amprenavir capsules) are available for oral administration. Each 50- mg capsule contains the inactive ingredients d-alpha tocopheryl polyethylene glycol 1000 succinate (TPGS), polyethylene glycol 400 (PEG 400) 246.7 mg, and propylene glycol 19 mg. The capsule shell contains the inactive ingredients d-sorbitol and sorbitans solution, gelatin, glycerin, and titanium dioxide. The soft gelatin capsules are printed with edible red ink. Each 50- mg AGENERASE Capsule contains 36.3 IU vitamin E in the form of TPGS. The total amount of vitamin E in the recommended daily adult dose of AGENERASE is 1,744 IU.
………………………………
paper
Org. Biomol. Chem., 2004,2, 2061-2070
DOI: 10.1039/B404071F
Efficient and industrially applicable synthetic processes for precursors of HIV protease inhibitors (Amprenavir, Fosamprenavir) are described. These involve a novel and economical method for the preparation of a key intermediate, (3S)-hydroxytetrahydrofuran, from L-malic acid. Three new approaches to the assembly of Amprenavir are also discussed. Of these, a synthetic route in which an (S)-tetrahydrofuranyloxy carbonyl is attached to L-phenylalanine appears to be the most promising manufacturing process, in that it offers satisfactory stereoselectivity in fewer steps.
……………

The reaction of N,N-dibenzyl-L-alaninal (I) with nitromethane, catalyzed by the chiral ammonium salt (II) and KF in THF gives the chiral nitroalcohol (III), which is reduced with NiCl2 and NaBH4 to yield the aminoalcohol (IV). The condensation of (IV) with isobutyraldehyde (V) affords the Schiff base (VI), which is reduced with NaBH4 to provide the secondary amine (VII). The reaction of (VII) with 4-nitrobenzenesulfonyl chloride (VIII) and TEA in dichloromethane furnishes the sulfonamide (IX), which is deprotected by hydrogenation with H2 over Pd/C in methanol, giving the diamino compound (X). Finally, this compound is condensed with 3(S)-tetrahydrofuryl (N-oxysuccinimidyl) carbonate (XI) by means of TEA in dichloromethane to afford the target carbamate.
Angew Chem. Int Ed Engl1999,38,(13-14):1931
……………………………………………………….
The reaction of the chiral epoxide (I) with isobutylamine (II) in refluxing ethanol gives the secondary amine (III), which is protected with benzyl chloroformate (IV) and TEA, yielding the dicarbamate (V). Selective deprotection of (V) with dry HCl in ethyl acetate affords the primary amine (VI), which is treated with 3(S)-tetrahydrofuryl N-succinimidinyl carbonate (VII) (prepared by condensation of tetrahydrofuran-3(S)-ol (VIII) with phosgene and N-hydroxysuccinimide (IX)) and DIEA in acetonitrile to provide the corresponding carbamate (X). The deprotection of (X) by hydrogenation with H2 over Pd/C in ethanol gives the secondary amine (XI), which is condensed with 4-nitrophenylsulfonyl chloride (XII) by means of NaHCO3 in dichloromethane/water to yield the sulfonamide (XIII). Finally, the nitro group of (XIII) is reduced with H2 over Pd/C in ethyl acetate to afford the target compound.
EP 0659181; EP 0885887; JP 1996501299; US 5585397; WO 9405639
……………………………………………………………
Patent
https://www.google.com/patents/WO1999048885A1?cl=ensynthesis of (3S)-tetrahydro-3-furyl N-[(1S,2R)-3(4-amino-N-isobutylbenzenesulphonamido)-1-benzyl-2-hydroxypropyl]carbamate, hereinafter referred to as the compound of formula (I), and to novel intermediates thereto.The compound of formula (I) has the following structure






 .
.
and was first described in PCT patent publication number WO94/05639 at Example 168. Currently there is considerable interest in the compound of formula (I) as a new chemotherapeutic compound in the treatment of human immunodeficiency virus (HIV) infection and the associated conditions such as acquired immune deficiency syndrome (AIDS) and AIDS dementia.
There exists at the present time a need to produce large quantities of the compound of formula (I) for clinical investigation into the efficacy and safety of the compound as a chemotherapeutic agent in the treatment of HIV infections.
An ideal route for the synthesis of the compound should produce the compound of formula (I) in high yields at a reasonable speed and at low cost with minimum waste materials and in a manner that is of minimum impact to the environment in terms of disposing of waste-materials and energy consumption.
We have found a new process for the synthesis of the compound of formula (I) with many advantages over previously known routes of synthesis. Such advantages include lower cost, less waste and more efficient use of materials. The new process enables advantageous preparation of the compound of formula (I) on a manufacturing scale.
The route of synthesis of the compound of formula (I) described in the specification of WO94/05639 is specifically described therein in examples 39A, 51A, 51B, 51C, 51D, 167 and 168. The overall yield from these examples is 33.2% of theory.
Generally the route described in WO94/05639 involves protecting the amino alcohol of formula (A) (Ex.39)
wherein P is a protecting group to form a compound of formula (B);
wherein P and P′ are each independently a protecting group;
deprotecting the compound of formula (B) to form a compound of formula (C) (Ex 51A);
wherein P′ is a protecting group;
forming a hydrochloride salt of compound (C) (Ex 51B) then reacting with N-imidazolyl-(S)-tetrahydrofuryl carbamate to form the compound of formula (D) (Ex 51C);
wherein P′ is a protecting group;
deprotecting the compound of formula (D) (Ex 51D) wherein P′ is a protecting group to form the compound of formula (D) wherein P′ is H (Ex 51E); and coupling the resultant secondary amine on the compound of formula (D) to a p-nitrophenylsulphonyl group to form a compound of formula (E) (Ex 167);
the resultant compound of formula (E) is then reduced to form the compound of formula (I) (Ex 168).
In summary, the process disclosed in WO94/05639 for producing the compound of formula (I) from the compound of formula (A) comprises 6 distinct stages:
1) protecting,
2) deprotecting,
3) reacting the resultant compound with an activated tetrahydrofuranol group,
4) deprotecting,
5) coupling with a p-nitrophenylsulfonyl group, and
6) reducing the resultant compound to form a compound of formula (I).
Applicants have now found a process by which the compound of formula (I) may be prepared on a manufacturing scale from the same starting intermediate, the compound of formula (A), in only 4 distinct stages instead of 6. In addition to the associated benefits of fewer stages, such as savings in time and cost, the improved process reduces the number of waste products formed. Furthermore, product may be obtained in a higher yield, of approximately 50% of theory
.
EXAMPLESExample 1
(1S,2R)-tert-butyl N-[1-benzyl-2-hydroxy-3-(isobutylamino)propyl]carbamate (127.77 g, 379.7 mmol) was heated in toluene (888 ml) to 80° C. and triethylamine (42.6 g, 417.8 mmol) added. The mixture was heated to 90° C. and a solution of p-nitrobenzene sulphonyl chloride (94.3 g, 425.4 mmol) in toluene (250 ml) was added over 30 minutes then stirred for a further 2 hours. The resultant solution of the nosylated intermediate {(1S,2R)-tert-butyl N-[1-benzyl-2-hydroxy-3-(N-isobutyl- 4-nitrobenzenesulphonamido)propyl]carbamate } was then cooled to 80° C. The solution was maintained at approximately 80° C., and concentrated hydrochloric acid (31.4 ml, 376.8 mmol) was added over 20 minutes. The mixture was heated to reflux (approx 86° C.) and maintained at this temperature for an hour then a further quantity of concentrated hydrochloric acid (26.4 ml, 316.8 mmol) was added. Solvent (water and toluene mixture) was removed from the reaction mixture by azeotropic distillation (total volume of solvent removed approx 600 ml), and the resultant suspension was cooled to 70-75° C. Denatured ethanol (600 ml) was added, and the solution was cooled to 20° C. The mixture was further cooled to approximately −10° C. and the precipitate formed was isolated by filtration, washed with denatured ethanol (50 ml) and dried at approximately 50° C., under vacuum, for approximately 12 hours, to give (2R,3S)-N-(3-amino-2-hydroxy-4-phenylbutyl)-N-isobutyl-4-nitrobenzene sulphonamide hydrochloride (160 g; 73% of theory yield corrected for assay). NMR: 1H NMR (300Mhz, dmso-d6): 8.37(2H, d, J=9 Hz), 8.16(NH3 +s), 8.06(2H, d, J=9 Hz), 7.31(5H, m), 5.65(1H, d, J=5 Hz), 3.95(1H, m), 3.39(2H, m), 2.95(5H, m), 1.90(1H, m), 0.77(6H, dd, J=21 Hz and 6 Hz).
1,1′-carbonyidiimidazole (27.66 kg, 170.58 mol) was added to ethyl acetate (314.3 kg) with stirring to give 3-(S)-tetrahydrofuryl imidazole-1-carboxylate. (S)-3-hydroxytetrahydrofuran (157 kg, 178.19 mol) was added over 30 minutes, washed in with ethyl acetate (9.95 kg), then the mixture was stirred for a further hour. (2R,3S)-N-(3-amino-2-hydroxy-4-phenylbutyl)-N-isobutyl-4-nitrobenzene sulphonamide hydrochloride (65.08 kg, 142.10 mol) was added and the mixture heated to reflux for approximately 22 hours. The solution was cooled slightly, and denatured ethanol (98 l) was added. The solution was stirred at 60° C. for 10 minutes then cooled and the product allowed to crystallise. The mixture was cooled to <10° C. and stirred for 2 hours. The product was isolated by filtration, washed with denatured ethanol (33 l) and dried at approximately 50° C., under vacuum to give (3S)-tetrahydro-3-furyl N-[(1S,2R)-1-benzyl-2-hydroxy-3-(N-isobutyl-4-nitrobenzene sulphonamido)propyl]carbamate in a yield of 82% of theory.
NMR: 1H NMR (500 Mhz, dmso-d6): 8.38(2H, d, J=9Hz), 8.06(2H, d, J=9 Hz), 7.20(6H, m), 5.02(1H, d, J=5 Hz), 4.94(1H, m), 4.35(EtOH, broad s), 3.71(EtOH, q), 3.65(1H, m), 3.60(1H, m), 3.51(2H, broad m), 3.40(2H, m), 3.15(1H, dd, J=8 Hz and 14 Hz), 3.07(1H, dd, J=8 Hz and 15 Hz), 2.94(2H, m), 2.48(1H, m), 2.06(1H, m), 1.97(1H, m), 1.78(1H, m), 1.05(EtOH, t), 0.83(6H, dd, J=7 Hz and 16 Hz).
Product from the above stage (80.0 g, 149.4 mmol) was hydrogenated in isopropanol (880 ml) with 5% palladium on carbon (16 g, of a wet paste) and hydrogen pressure (approx 0.5 to 1.5 bar) at 25-50° C. for approximately 5 hours. The mixture was cooled and the catalyst removed by filtration. The solution was distilled to a volume of approximately 320 ml and water (80 ml) was added. This solution was divided into two for the crystallisation step.
To half of the above solution, decolourising charcoal (2 g) was added, the mixture stirred at approximately 32° C. for 4 hours, then filtered. The filtercake was washed with isopropanol (20 ml) then further water (40 ml) was added to the filtrate. The solution was seeded to induce crystallisation and stirred for 5 hours. Water (130 ml) was added slowly over 1 hour then the mixture was stirred for 4 hours. The resultant slurry was cooled to approximately 20° C. and the product was isolated by filtration and washed with a 1:4 mixture of isopropano/water (120 ml). The product was dried at approximately 50° C., under vacuum, for approximately 12 hours to give (3S)-tetrahydro-3-furyl N-[(1S,2R)-3-(4-amino-N-isobutylbenzenesulphonamido)-1-benzyl-2-hydroxypropyl] carbamate (30.3 g; 80% of theory yield).
NMR: 1H NMR (300 Mhz, dmso-d6): 7.39(2H, d, J=9 Hz), 7.18(6H, m), 6.60(2H, d, J=9 Hz), 6.00(2H, s), 4.99(1H, d, J=6 Hz), 4.93(1H, ddt), 3.64(5H, m), 3.34(1H, m), 3.28(1H, dd, J=14 Hz and 3 Hz), 3.01(1H, m, J=14 Hz and 3 Hz), 2.91(1H, m), 2.66(2H, m), 2.50(1H, m), 2.05(1H, m), 1.94(1H, m), 1.78(1H, m), 0.81(6H, dd, J=16 Hz and 7 Hz). m/z: 506.2(M+H+)
…………………………
PATENT
Example 11Synthesis of Amprenavir (1)To a solution of carbamate nitro derivative 15 (0.05 g, 0.09 mmol) in 2 mL of EtOAc was added SnCl2.2H2O (0.1 g, 0.5 mmol) at 70° C. The reaction mixture was heated for 1 h until starting material disappeared and the solution cooled to room temperature. It was then poured into saturated aq. NaHCO3 solution and extracted with EtOAc. The organic extract was dried over anhyd. Na2SO4 and concentrated under reduced pressure. It was purified over chromatography using petroleum ether:EtOAc (3:2) to give amprenavir 1 (0.04 g, 90%).IR: (CHCl3, cm−1): υmax757, 1090, 1149, 1316, 1504, 1597, 1633, 1705, 2960, 3371; 1H NMR (200 MHz, CDC3): δ 0.86 (d, J=5.7 Hz, 3H), 0.90 (d, J=6.6 Hz, 3H), 1.78-2.21 (m, 3H), 235-3.11 (m, 6H), 3.58-4.11 (m, 7H), 4.25 (s, 2H), 5.01 (br s, 1H), 5.07 (br s, 1H), 6.65 (d, J=8.4 Hz, 2H), 7.20-7.28 (m, 5H), 7.51 (d, J=8.4 Hz, 2H);13C NMR (50 MHz, CDC3): δ 19.9, 20.2, 27.3, 32.8, 35.4, 35.7, 53.8, 55.0, 58.6, 66.8, 72.6, 73.2, 75.3, 114.0, 125.9, 126.5, 1280.4, 129.5, 137.7, 150.9, 155.9;
Anal. Calcd for C25H35N3O6S: C, 59.39; H, 6.98; N, 8.31; S, 6.34. Found: C, 59.36; H, 6.81; N, 8.25; S, 6.29%.
……………………..
NMR PREDICTIONS
1H NMR
13 C NMR
COSY PREDICTION

See also
- Fosamprenavir, a prodrug of amprenavir
External links
- Amprenavir bound to proteins in the PDB
- Shen, C. H.; Wang, Y. F.; Kovalevsky, A. Y.; Harrison, R. W.; Weber, I. T. (2010). “Amprenavir complexes with HIV-1 protease and its drug-resistant mutants altering hydrophobic clusters”. FEBS Journal 277 (18): 3699–3714. doi:10.1111/j.1742-4658.2010.07771.x. PMC 2975871.PMID 20695887.
 | |
| SYSTEMATIC (IUPAC) NAME | |
|---|---|
| (3S)-oxolan-3-yl N-[(2S,3R)-3-hydroxy-4-[N-(2-methylpropyl)(4-aminobenzene)sulfonamido]-1-phenylbutan-2-yl]carbamate | |
| CLINICAL DATA | |
| TRADE NAMES | Agenerase |
| AHFS/DRUGS.COM | monograph |
| MEDLINEPLUS | a699051 |
| LICENCE DATA | EMA:Link, US FDA:link |
| |
| LEGAL STATUS |
?
|
| ROUTES | oral |
| PHARMACOKINETIC DATA | |
| PROTEIN BINDING | 90% |
| METABOLISM | hepatic |
| HALF-LIFE | 7.1-10.6 hours |
| EXCRETION | <3% renal |
| IDENTIFIERS | |
| CAS NUMBER | 161814-49-9 |
| ATC CODE | J05AE05 |
| PUBCHEM | CID 65016 |
| DRUGBANK | DB00701 |
| CHEMSPIDER | 58532 |
| UNII | 5S0W860XNR |
| KEGG | D00894 |
| CHEBI | CHEBI:40050 |
| CHEMBL | CHEMBL116 |
| NIAID CHEMDB | 006080 |
| CHEMICAL DATA | |
| FORMULA | C25H35N3O6S |
| MOLECULAR MASS | 505.628 g/mol |

No comments:
Post a Comment