
SET 1/3 IS AT http://drugsynthesisint.blogspot.in/p/tinib-series.html
TINIB SERIES 1/3
1 IBRUTINIB
2.COBIMETINIB
3.AFATINIB
4 SELUMETINIB
5 ALECTINIB
6 IMATINIB
7 NERATINIB
8 ERLOTINIB
9 AMUVATINIB
SET 2/3 STARTS HERE
TINIB SERIES CONT 2/3
10 BINIMETINIB
11 DACOMITINIB
12 MOMELOTINIB
13 PALBOCICLIB
14 SORAFENIB
15 NINTEDANIB
16 LAPATINIB
17 QUIZARTINIB
18 BOSUTINIB
19
10 BINIMETINIB

Binimetinib
5-[(4-bromo-2-fluorophenyl)amino]-4-fluoro-N-(2-hydroxyethoxy)-1-methyl-1H-benzimidazole-6-carboxamide
5-(4-Bromo-2-fluorophenylamino)-4-fluoro-1-methyl-1H-benzimidazole-6-carbohydroxamic acid 2-hydroxyethyl ester
6-(4-Bromo-2-fluorophenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid (2-hydroxyethyoxy)-amide
606143-89-9 CAS
C17H15BrF2N4O3, 441.227
tyrosine kinase inhibitor, antineoplastic
Array BioPharma Inc;PHASE 3 Cancer, ovary (serous)
Novartis PHASE 3 Melanoma
AGARRY-162
ARRY-438162
MEK-162
ARRY-438162
MEK-162
MEK-1 protein kinase inhibitor; MEK-2 protein kinase inhibitor
Liver injury; Melanoma; Noonan syndrome; Ovary tumor; Solid tumor
Growth factor-mediated proliferative signals are transmitted from the extracellular environment to the nucleus through several pathways, including the RAS/RAF/ MEK pathway. The RAS/RAF/MEK kinase signal transduction pathway is activated through initial extracellular binding and stimulation of tyrosine receptor kinases (RTKs) by their respective cognate ligands. Upon autophosphorylation of specific tyrosine residues in the cytosolic domain of RTKs, the Grb2-Sos complex translocates to the plasma membrane, and converts the inactive RAS'GDP to active RAS'GTP. The interaction between the Grb2 docking protein and the activated kinases or the phosphorylated receptor associated proteins is mediated by the Src Homology (SH2) domain of the signaling protein that recognizes specific phosphotyrosine sequences. RAS undergoes a conformational change upon guanosine 5 '-triphosphate (GTP) binding and causes the recruitment of RAF- 1 to the cytoplasmic membrane where it is phosphorylated by several kinases and simultaneous disphosphorylated at key residues by protein phosphatase-2B. Activated RAF phosphorylates the mitogen- activated protein kinase kinase (MEK) on two serine residues in the activation loop, which results in the activation of this protein kinase. MEK then phosphorylates and activates extracellular signal-regulated kinase (ERK), allowing its translocation to the nucleus where it phosphorylates transcriptional factors permitting the expression of a variety of genes.
The RAS/RAF/MEK signal transduction pathway is deregulated, often through mutations that result in ectopic protein activation, in roughly 1/3 of human cancers. This deregulation in turn results in a wide array of cellular changes that are integral to the etiology and maintenance of a cancerous phenotype including, but not limited to, the promotion of proliferation and evasion of apoptosis (Dhillon et al., Oncogene, 2007, 26: 3279-3290).
Accordingly, the development of small molecule inhibitors of key members of the RAS/ RAF/ MEK signal transduction pathway has been the subject of intense effort within the pharmaceutical industry and oncology community.
MEK is a major protein in the RAS/ RAF/ MEK pathway, which signals toward cell proliferation and survival, and frequently activated in tumors that have mutations in the RAS or RAF oncogenes or in growth receptor tyrosine kinases. MEK is a key player in the RAS/RAF/MEK pathway as it is downstream of RAS and RAF. Despite being only rarely mutated in cancer (Murugan et al., Cell Cycle, 2009, 8: 2122-2124; Sasaki et al., J. Thorac. Oncol., 2010, 5: 597-600), inhibitors of the MEK1 and MEK2 proteins have also been targeted for small molecule inhibition owing to their central position within the RAS/ RAF/ MEK signal transduction pathway signaling cascade (Fremin and Meloche, J. Hematol.
Oncol., 2010, 3:8). Recently a potent MEK inhibitor failed to demonstrate efficacy in clinical trials in patients with advanced non-small cell lung cancer (Haura et al., Clin. Cancer Res., 2010, 16: 2450-2457). The reason for failure in this trial is not clear.
6-(4-Bromo-2-fluorophenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid (2-hydroxyethyoxy)-amide (hereinafter, "Compound A") is a benzimidazole compound that is a known potent and selective inhibitor of the MEK1 and MEK2 proteins, and useful in the treatment of hyperproliferative diseases, particularly cancer, in mammals. For example, in a recently published Phase I study of 28 patients suffering from unresectable, locally advanced or metastatic biliary cancer and who had received < 1 prior systemic therapy, oral Compound A treatment (60 mg twice daily) resulted in 1 complete regression, 1 partial regression and 11 stable disease diagnoses after at least 6 weeks of treatment (Finn et al., J. Clin. Oncol. 30, 2012 (Supplement 4, 2012 Gastrointestinal Cancers Symposium, Abstract No. 220). Compound A has also been demonstrated to be effective in the treatment of patients with either BRAFV600 or NRAS-mutant melanoma (Ascierto et al., J. Clin. Oncol. 30, 2012 (Supplement, 2012 ASCO Annual Meeting, Abstract No. 8511).
The compound, as well as a process for its preparation, is disclosed in PCT Pub. No. WO 03/077914
MEK-162, a potent, orally active MEK1/2 inhibitor, is in phase III clinical trials at Array BioPharma and licensee Novartis for the treatment of metastatic or unresectable cutaneous melanoma with NRAS mutations and in combination with LGX-818 in adult patients with BRAF V600. Phase III studies are also under way at Array BioPharma for the treatment of low grade serous carcinomas of the ovary, fallopian tube or primary peritoneum following at least one prior platinum-based chemotherapy regimen and no more than three lines of prior chemotherapy regimens. Novartis and Array BioPharma are also conducting phase II clinical studies for the treatment of locally advanced and unresectable or metastatic malignant cutaneous melanoma, harboring BRAFV600E mutations; in BRAF mutated melanoma in combination with AMG-479 and for the treatment of Noonan's syndrome, and in non-small cell lung cancer harboring KRAS or EGFR mutation and in combination with erlotinib. MEK-162 is being evaluated in phase I/II as first line treatment of advanced biliary tract carcinoma and for the treatment of adult patients with mutant or wild-type RAS metastatic colorectal cancer. The product is in early clinical trials at Array Biopharma for the treatment of biliary cancer.
According to Array, MEK-162 may also provide broad therapeutic benefits in the treatment of chronic degenerative diseases. However, a phase II trial for the treatment of stable rheumatoid arthritis (RA) did not meet its primary endpoint. Based on these data, the company focused development of MEK-162 solely in oncology.
In 2010, MEK-162 was licensed to Novartis by Array BioPharma for worldwide development. In 2013, orphan drug designation was assigned in Japan for the treatment of malignant melanoma with NRAS or BRAF V600 mutation.
WO-2014063024 DEALS WITH Preparation, crystalline forms, and formulations comprising binimetinib. Binimetinib is a MEK-1/2 inhibitor originally claimed in WO03077914, which Array and Novartis are developing for the treatment of cancer, including melanoma, low-grade serous ovarian cancer, and other solid tumors, as well as Noonan syndrome hypertrophic cardiomyopathy and hepatic impairment. See also WO2014018725 for the most recent filing on the agent
//////////////////////////
WO 03/077914
Schemes 1-4.
Scheme 1


Scheme la

Scheme 2

Scheme 3

17 18
Scheme 4

25
Scheme 5


General synthetic methods which may be referred to for preparing some of the compounds of the present invention are provided in PCT published application number WO 00/42022 (published July 20, 2000). The foregoing patent application is incorporated herein by reference in its entirety.
similar ie chloro instead of fluoro
Example 52

6-(4-Bromo-2-chloro-phenylamino)-7-chloro-3-methyl-3H-benzoimidazole-5- carboxylic acid (2-hydroxy-ethoxy)-amide (lOcc) Step A: 3-Chloro-2,4-difluoro-5-nitro-benzoic acid 2a
3-Chloro-2,4-difluoro-benzoic acid la (3.00 g, 15.6 mmol) is added to a stirred solution of concentrated H2SO4 (16 mL) and fuming nitric acid (0.85 mL, 20.3 mmol). After 3 hours a precipitate forms. The yellow slurry is poured onto ice water (100 mL). The aqueous mixture is extracted with diethyl ether (3x). The organic extracts are dried (Na2SO4) and concentrated under reduced pressure to give 3.50 g (95%) of clean desired product as a pale yellow solid.
Step B: 4-Amino-3-chloro-2-fluoro-5-nitro-benzoic acid 3a
Ammonium hydroxide solution (6.88 g, -30% in water, 58.9 mmol) is added to a solution of 3-chloro-2,4-difluoro-5-nitro-benzoic acid 2a (3.5 g, 14.7 mmol) in water (16 mL) at 0 °C with stirring. Upon completion of the ammonium hydroxide addition the reaction mixture is warmed to room temperature. After 5 hours the reaction mixture is cooled to 0 °C and concentrated HCl is carefully added until the pH of the reaction mixture is near zero. The solid is collected by filtration and washed with water and diethyl ether. The solids are transferred to a round bottom flask as a solution in MeOH and EtOAc and concentrated under reduced pressure to give 2.96 g of a yellow solid. The filtrate is partitioned between diethyl ether and water and the organic layer is washed with brine. The combined organic extracts are dried (Na2SO ) and concentrated under reduced pressure to give 0.65 g of product. Recovered a total of 3.61 g (104%) of pure desired product, that is carried forward without further purification.
Step C: 4~Amino-3-chloro-2-fluoro-5-nitro-benzoic acid methyl ester 4a
To a stirred solution of 4-amino-3-chloro-2-fluoro-5-nitro-benzoic acid 3a (3.61 g, 15.4 mmol) in THF (30 mL) and MeOH (10 mL), TMS diazomethane (9.23 mL, 2.0 M solution in hexanes, 18.5 mmol) is added. After completion of reaction, the reaction mixture is concentrated via rotary evaporation with acetic acid in the trap. The recovered oily solid is triturated with diethyl ether to provide 1.51 g of a yellow solid. The filtrate is concentrated and triturated with diethyl ether to give an additional 0.69 g of yellow solid. A total of 2.20 g (57%) of pure desired product is recovered.
Step D: 4-Amino-3-chloro-5-nitro-2-phenylamino-benzoic acid methyl ester 5c
4-Amino-3-chloro-2-fluoro-5-nitro-benzoic acid methyl ester 4a (2.20 g, 8.84 mmol) is suspended in MeOH (9.4 mL) and aniline (3.22 mL, 35.4 mmol) is added. The reaction mixture is heated to reflux with stirring under a nitrogen atmosphere. After 19 hours, the reaction is complete. Distilled water (3.22 mL) is added to the reaction mixture and refluxing is continued for one hour. The reaction mixture is cooled to 0 °C in an ice bath for 20 minutes. The reaction mixture is filtered and washed with 3:10 distilled water/MeOH (65 mL total) and then with MeOH. The solid is dissolved with CH2C12 and concentrated under reduced pressure to give 2.40 g (84%) of pure desired product. MS APCI (-) m/z 320.3 (M-l) detected.
Step E: 4, 5-Diamino-3-chloro-2-phenylamino-benzoic acid methyl ester 6b
4-Amino-3-chloro-5-nitro-2-phenylamino-benzoic acid methyl ester 5c (0.50 g, 1.55 mmol) is dissolved into 2:1 EtOH/MeOH (15.5 mL). Saturated aqueous NH4C1 (15 mL), Zn powder (1.02 g, 15.6 mmol), and THF (10 mL) are added. After stirring for 20 hours, the reaction mixture is diluted with CH C12/THF and water. The organic layer is washed with water (3x). The combined organic extracts are dried (Na2SO4) and concentrated under reduced pressure. The solids are triturated with ether to give 0.32 g (70%) clean desired product. Step F: 7-Chloro-6-phenylamino-3H-benzoimidazole-5-carboxylic acid methyl ester 7c
4,5-Diamino-3-chloro-2-phenylamino-benzoic acid methyl ester 6b (0.32 g, 1.09 mmol) and formamidine acetate (72 mg, 1.64 mmol) in EtOH (36 mL) are heated, with stirring, to 80 °C. After 44 hours, the reaction mixture is cooled to room temperature and diluted with EtOAc and washed with water (3x), saturated NaHCO3, and brine. The combined organic extracts are dried (Na2SO4) and concentrated under reduced pressure to give 0.33 g (99%) clean desired product as a solid. MS APCI (+) m/z 302.3 (M+l) detected.
Step G: 6-(4-Bromo-phenylamino)-7-chloro-3H-benzoimidazole-5-carboxylic acid methyl ester 8g
7-Chloro-6-phenylamino-3H-benzoimidazole-5-carboxylic acid methyl ester 7c (0.327 g, 1.08 mmol) is dissolved into DMF (16 mL) and NBS (0.193 g, 1.08 mmol) is added. After one hour, the reaction mixture is quenched by the addition of saturated aqueous NaHSO3. The reaction mixture is then partitioned between EtOAc/THF and water. The organic layer is washed with water and brine. The combined organic extracts are dried (Na2SO ) and concentrated under reduced pressure. The recovered solid is triturated with ether to give 0.225 g (54%) pure desired product. MS ESI (+) m/z 382, 384 (M+, Br pattern) detected.
Step H: 6-(4-Bromo-2-chloro-phenylamino)- 7 -chloro-3H-benzoimidazole-5 -carboxylic acid methyl ester lOdd 6-(4-Bromo-phenylamino)-7-chloro-3H-benzoimidazole-5-carboxylic acid methyl ester 8g (0.225 g, 0.591 mmol) is dissolved in DMF (2 mL) and NCS (79 mg, 0.591 mmol) is added. After the NCS is in solution concentrated HCl (0.005 mL, 0.059 mmol) is added. After 2 hours, sodium bicarbonate, water and NaHSO3 are added to the reaction mixture. Solids are filtered and washed with water and ether to give 0.141 g (57%) of clean desired product as a tan solid. MS APCI (-) m/z 414, 416 (M-, Br pattern) detected.
Step I: 6-(4-Bromo-2-chloro-phenylamino)-7-chloro-3-methyl-3H-benzoimidazole-5- carboxylic acid methyl ester lOee
6-(4-Bromo-2-chloro-phenylamino)-7-chloro-3H-benzoimidazole-5-carboxylic acid methyl ester lOdd (0.141 g, 0.34 mmol), potassium carbonate (0.141 g, 1.02 mmol), and iodomethane (0.063 mL, 1.02 mmol) are dissolved in dimethylformamide (3 mL). After 20 hours, the reaction mixture is diluted with EtOAc and washed with water (3x), potassium carbonate, and brine. The organic layer is dried (Na2SO4) and concentrated to a brown oil. The N3 and Nl alkylated regioisomers are separated by flash chromatography (EtOAc). The recovery of the N3 alkylated regioisomer is 20.4 mg (28%). MS ESI (+) m/z 428, 430 (M+, Br pattern) detected.
Step J: 6-(4-Bromo-2-chloro-phenylamino)-7-chloro-3-methyl-3H-benzoimidazole-5- carboxylic acid 10 ff
6-(4-Bromo-2-chloro-phenylamino)-7-chloro-3-methyl-3H-benzoimidazole-5- carboxylic acid methyl ester lOee (21 mg, 0.048 mmol) is dissolved into 2:1 THF/water (1.2 mL) and NaOH (0.190 mL, 1.0 M aqueous solution, 0.190 mmol) is added. After stirring for 4 hours the reaction is diluted with water and acidified to pH 2 by addition of 1.0 M HCl. The mixture is then extracted with 3:1 EtOAc/THF (3x), dried (Na2SO ) and concentrated to give quantitative yield of desired prodcut as a white solid. MS APCI (+) m/z 414, 416 (M+, Br pattern) detected.
Step K: 6-(4-Bromo-2'chloro-phenylamino)- 7-chloro-3-methyl-3H-benzoimidazole-5- carboxylic acid (2-vinyloxy-ethoxy) -amide lOgg
6-(4-Bromo-2-chloro-phenylamino)-7-chloro-3-methyl-3H-benzoimidazole-5- carboxylic acid lOff (32 mg, 0.077 mmol), O-(2-vinyloxy-ethyl)-hydroxylamine (0.010 mL, 0.092 mmol), HOBt (13 mg, 0.093 mmol), triethylamine (0.011 mL, 0.077 mmol), and EDCI (19 mg, 0.10 mmol) are dissolved into dimethylformamide (1.0 mL) and allowed to stir under a nitrogen atmosphere at room temperature for 24 hours. The reaction mixture is diluted with EtOAc, washed with water (3x), 10% potassium carbonate (2x), saturated ammonium chloride, brine, dried (Na2SO4), and concentrated under reduced pressure to give 39 mg of 85% pure material. MS APCI (-) m/z 497, 501 (M-, Br pattern) detected.
Step L: 6-(4-Bromo-2-chloro-phenylamino)-7-chloro-3-methyl-3H-benzoimidazole-5- carboxylic acid (2-hydroxy-ethoxy)-amide lOcc
Hydrochloric acid (0.78 mL, 1.0 M aqueous solution, 0.78 mmol) is added to a suspension of 6-(4-bromo-2-chloro-phenylamino)-7-chloro-3-methyl-3H- benzoimidazole-5-carboxylic acid lOgg (2-vinyloxy-ethoxy)-amide (39 mg, 0.078 mmol) in MeOH (1 mL). After one hour, the reaction mixture is neutralized to pH 7 and concentrated under reduced pressure. The solids are dissolved in EtOAc, washed with brine, dried (Na SO4), and concentrated under reduced pressure. Flash chromatography (20:1 CH2Cl2/MeOH) provides 9 mg (23%) of pure product: MS APCI (+) m/z 473, 475 (M+, Br pattern) detected; 1H NMR (400 MHz, CDC13) δ 8.30 (s, IH), 8.08 (s, IH), 7.57
(d, IH), 7.15 (dd, IH), 6.21 (d, IH), 3.97 (s, 3H) 3.86 (m, 2H), 3.57 (m, 2H).
actual is below
Example 18
The following compounds are prepared by methods similar to those described in
Example 10 by using methyl ester 8d and the appropriate alkylating agent (Step A) and
the appropriate hydroxylamine (Step C):
/////////////////////
COMPD A
Example 1. Preparation of 6-(4-Bromo-2-fluorophenylamino)-7-fluoro-3-methyl-3H-

Compound 1 Compound 3
In an inertized (N2) reaction vessel at internal temperature 20°C and under exclusion of humidity and air, Compound 1 (1.0 eq.) and Compound 2 (1.2 eq.) are reacted in the presence of cesium carbonate (2.4 eq.), tris(dibenzylidenaceton) dipalladium(O) (0.035 eq.) and Xantphos (0.07 eq.) in a mixture of toluene and 1 ,4-dioxane at internal temperature of 99°C. After 8 hours, the mixture is cooled to internal temperature of 60°C.
Subsequently, dimethylformamide (DMF), filter aid (CEFOK) and activated charcoal (EKNS) are added, and the mixture is stirred and cooled to internal temperature of 35 °C. The solids are filtered off and washed with a mixture of dimethylformamide and toluene. To the filtrate, which contains the product Compound 3, is introduced at internal temperature of
25 °C hydrogen chloride gas (CLC) whereupon the HQ salt of Compound 3 crystallizes. The palladium residue mainly remains in solution. After warming to 60 °C and cooling to 0°C, the solids are filtered using a centrifuge and are washed with a mixture of toluene and dimethylformamide.
The damp Compound 3 HC1 salt is charged to a reactor (equipped with pH probe) together with dimethylformamide and is heated to 60°C. By adding a 4 wt% of aqueous tripotassium phosphate solution, the pH is adjusted to a pH range of 6.8-7.6 (with a target of pH 7.2) while Compound 3 crystallizes as free base. After cooling to 22°C and stirring, the solids are filtered using a centrifuge and are washed with drinking water. The moist solids are dried at 50 °C under vacuum to give dry, crude Compound 3.
In order to remove residual palladium, dry, crude Compound 3 is dissolved in dimethylformamide at internal temperature of 60°C and stirred together with Smopex-234 (commercially available from Johnson Matthey) and activated charcoal for 90 minutes. The solids are filtered off at internal temperature of 60°C and are washed with
dimethylformamide. To the filtrate are added drinking water and Compound 3 seed crystals. More drinking water is added while Compound 3 crystallizes. After cooling to internal temperature of 20 °C, the solids are filtered using a centrifuge and are washed with a mixture of deionized water and dimethylformamide and with deionized water. The moist solids are dried at 50°C under vacuum, providing 6-(4-Bromo-2-fluorophenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid methyl ester (Compound 3).
Example 2. Preparation of 6-(4-Bromo-2-fluorophenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid-(2-tert-butoxyethoxy)-amide
A. "One-pot" Synthesis

Compound 3 Intermediate 1
t-Bu-O. /\ ^ H2
(Compound 4)

Compound 5
In an inertized reaction vessel at internal temperature 20-25 °C under nitrogen, 6-(4-Bromo-2-fluorophenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid methyl ester (Compound 3, 1.0 eq.) is added to a mixture of DMF and THF. To this slurry, a solution of potassium trimethylsilanolate (1.05 eq.) in THF is added to the mixture at internal temperature of 25 °C over a period of about 40 minutes, and the resulting mixture is stirred for about 1 hour, providing a potassium salt solution of Intermediate 1. A THF/methanol mixture is then sequentially distilled off from the mixture at 85-120°C during about 2 hours.
The potassium salt solution is then added to a suspension of CDI (1.25 eq.) and imidazole hydrochloride (1.40 eq.) in THF at internal temperature of 25 °C over a period of about 1 hour. The resulting mixture is then stirred for approximately 1 hour at 50°C, and the following imidazolide intermediate

The imidazolide intermediate is not further isolated.
Subsequently, 1.2 eq. of 0-(2-tert-butoxyethyl)hydroxylamine (Compound 4, CAS No. 1023742-13-3, available from suppliers such as Huhu Technology, Inc.®) is added over a period of about 30 minutes at 50°C and stirred for 1.5 hours. Demineralized water is then added at 50°C, producing a precipitate. After cooling to 20°C and stirring for about 3-16 hours, the slurry is filtered off, washed with THF/ demineralized water (1 :2) in 2 portions and with demineralized water in three portions, and dried at 50°C / <70 mbar for about 17 hours, providing 6-(4-bromo-2-fluorophenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid-(2-tert-butoxyethoxy)-amide (Compound 5) as monohydrate.
B. A synthesis method with isolation of the intermediate of step a) from the reaction mixture of step a) prior to the reaction of step b)
Alternatively, 6-(4-bromo-2-fluorophenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5 -carboxylic acid-(2-tert-butoxyethoxy)-amide (Compound 5) can be made by the synthesis method as shown below. Compound 3, which is a methyl ester, is first converted to a carboxylic acid, which is then isolated by a crystallization to form Compound
6. Compound 6 is then coupled with Compound 4 to form Compound 5 as monohydrate.
The crystallization step in this method removes starting materials such as Compound 1, process impurities, and the dba ligand from the prior catalyst before the coupling reaction with Compound 4, and at the same time maintains the overall yield of the synthesis.


6-(4-Bromo-2-fluorophenylamino)-7-fluoro-3-memy acid In an inertized (N2) reaction vessel at internal temperature of 60°C, Compound 3 (1.0 eq.) is dissolved in DMF and stirred with a fiber, which is sold under the trademark
SMOPEX 234, and activated charcoal for the removal of palladium to not more than 100 ppm. The fiber and activated charcoal are removed by filtration at 60°C and washed with DMF.
The filtrate (containing Compound 3) is transferred to a second inertized (N2) reaction vessel and cooled to an internal temperature of 30°C. A thin suspension can form at this point of time. 30% sodium hydroxide (1.1 eq.) and water (for rinsing) are added, and the resulting reaction mixture is vigorously stirred for 3 hours at an internal temperature of 30 °C. The methyl ester is saponified. Conversion is checked by an IPC (HPLC). As soon as the IPC criterion is met, a filter aid, which is sold under the trademark HYFLO, is added. The mixture is stirred for 15 minutes and then filtered at 30°C via a plate filter and polish filter to a third reaction inertized (N2) vessel.
An aqueous HC1 solution 7.5 % is added to the clear filtrate in the third vessel at an internal temperature of 30 °C until a pH value of 8 is reached. Then the solution is seeded at an internal temperature of 30°C with Compound 6, and an aqueous HC1 solution 7.5 % is added under vigorous stirring until a pH value of pH 2.8 is reached. The product gradually crystalizes. The suspension is cooled over 60 min to an internal temperature of 25 °C and
water is added. The suspension is stirred for at least 4 hours at an internal temperature of 25°C.
The resulting solid is collected by centrifugation or filtration. The filter cake is first washed with DMF/water 1 :1 (w/w) and then with water, discharged and dried in a vacuum at 50°C. The water content is controlled by IPC. The crystalline product Compound 6 is discharged as soon as the IPC criterion is met.
6-(4-Bromo-2-fluorophenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid- (2-tert-butoxyethoxy) - amide
An inertized (N2) reaction vessel is charged with Compound 6 (1.0 eq.), DMF, and
THF at room temperature. The suspension is heated to 25 °C under stirring with flow of nitrogen. After CDI (1.13 eq.) is added, the suspension can get thinner and slight evolution of gases can be observed. After the suspension finally becomes a solution, it is then monitored by IPC (HPLC).
As soon as the IPC (HPLC) criterion is met, the reaction mixture is heated to 50°C over 20 minutes and imidazole hydrochloride (0.3 eq.) is added, forming a solution of
Intermediate 2.
To the solution of Intermediate 2, Compound 4 (1.3 eq.) is added over 60 minutes at internal temperature of 50°C under stirring at a speed of 300 rpm with flow of nitrogen. As soon as the IPC (HPLC) criterion is met, the mixture is cooled to 20-25 °C over 30 minutes. The mixture is then stored at ambient temperature overnight under nitrogen without stirring. DMF is added to the mixture followed by heating it to 50 °C over 30 minutes. Complete conversion of Intermediate 2 to Compound 5 is confirmed by IPC (HPLC).
Water is added to the mixture at internal temperature of 50 °C over 20 minutes. Then the solution is seeded with Compound 5. After stirring at 50 °C for 60 minutes, more water is added to the suspension at 50 °C over 90 minutes. After vigorous stirring, the suspension is cooled to 20 °C over 2 hours and filtered. The filter cake is washed twice with THF/water (v/v: 1 :2) at 20 °C, and twice with water at 20 °C. Finally, the filter cake is dried at 50 °C under vacuum to provide 6-(4-bromo-2-fluorophenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid-(2-tert-butoxyethoxy)-amide (Compound 5) as monohydrate.
Example 3. Preparation of 6-(4-Bromo-2-fluorophenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid (2-hydroxyethyoxy)-amide (Compound A)

Compound 5 Compound A
6-(4-Bromo-2-fluorophenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid-(2-tert-butoxyethoxy)-amide (Compound 5) monohydrate is added in 3 portions to a premixed solution of Acetonitrile and excess Phosphoric acid (85 % aqueous solution) at internal temperature 20-25 °C. After stirring for about 15 minutes, the suspension is heated to internal temperature 50-53 °C. The suspension is maintained at this temperature for 6 hours, cooled to internal temperature 20-25 °C. The mixture is then heated to internal temperature 35-37°C and diluted with Ethanol- Water (3 :1 v/v). EKNS and CEFOK are added, the reaction mixture is stirred approximately 15 minutes and filtered over a funnel coated with CEFOK. The filtrate is cooled to approximately 30°C. 3 N aqueous potassium hydroxide (ΚΟΗ) is added to the cooled filtrate over a period of 90 minutes until a pH- value of about 8.1 is reached. The suspension is heated to internal temperature 60-63 °C, stirred at this temperature for a period of about 2 hours, cooled to 20-23 °C over a period of about 45 minutes, filtered over a funnel, and dried at 50°C pressure <100 mbar over a period of about 17 hours, providing 6-(4-bromo-2-fluorophenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid (2-hydroxyethyoxy)-amide (Compound A) as a white powder.
Example 4. Preparation of Crystallized 6-(4-bromo-2-fluorophenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid (2-hydroxyethyoxy)-amide (Compound A) In a dry vessel at room temperature, Compound A is added to a premixed solvent solution of methanol/THF/water (35/35/30 w/w). The suspension is heated to internal temperature 53-55°C, and the resulting solution is hot filtered by deep and membrane filtration (via a paper filter and PTFE membrane) at internal temperature 53-56°C. The clear solution is stirred and cooled to 47-48°C, and the seed crystals suspension (i.e., seed crystals of crystallized Compound A in water, 10% m/m) is added (0.2 to 0.5% of crystallized Compound A expected yield mass). After about 20 minutes, water is slowly added within 25 hours (33.3% within 15 hours and 66.6% within 10 hours with at least 10 minute stirring after addition of water) to obtain a final ratio of methanol THF/water (20/20/60 w/w). After the water is added, the suspension is cooled down to internal temperature 3-5 °C within 10 hours and stirred for 0.5 hours. The white suspension is filtered over a sinter glass nutsche (75 ml, diameter = 6 cm, pore 3) suction filter and washed once with ice cold methanol/THF/water (15/15/70 w/w at 2-4 °C), and two times with ice cold water (2-4 °C). Drying takes place in a vacuum oven dryer at 20°C for 10 hours, and then at 40°C for 10 hours, and then at 60°C for at least 12 hours with pressure < lOmbar, providing crystallized Compound A.
Example 5. Pharmaceutical Composition
Crystallized Compound A is formulated as indicated in Table 1 :
Table 1

* The weight of the drug substance is taken with reference to the dried substance (100%) on the basis of assayed value. The difference in weight is adjusted by the amount of lactose monohydrate.
** The Opadry II is combined with the sterile water to make a 12% w/w Opadry II (85F) film coat suspension, which is then sprayed onto the core tablet.
*** Removed during processing
Upon mixing of the tablet core components, the pharmaceutical composition is converted into a tablet form by direct compression. The formed tablet may be further coated with the tablet coating provided above.
11
DACOMITINIB

Dacomitinib
(2E)-N-{4-[(3-Chloro-4-fluorophenyl)amino]-7-methoxy-6-quinazolinyl}-4-(1-piperidinyl)-2-butenamide
4-Piperidin-1-yl-but-2-enoic acid [4-(3-chloro-4-fluoro-phenylamino)-7-methoxy-quinazolin-6-yl]-amide
4-Piperidin-1-yl-but-2-enoic acid [4-(3-chloro-4-fluoro-phenylamino)-7-methoxy-quinazolin-6-yl]-amide
pf299804…… pfizer
EGFR (HER1; erbB1) Inhibitors
HER4 (erbB4) Inhibitors
HER2 (erbB2) Inhibitors
HER4 (erbB4) Inhibitors
HER2 (erbB2) Inhibitors
- Molecular formula:C24H25ClFN5O2
- Molecular mass:469.95
Dacomitinib (PF-00299804) is an experimental drug candidate under development by Pfizer for the treatment of non-small-cell lung carcinoma. It is a selective and irreversible inhibitor of EGFR.[1]
Dacomitinib has advanced to several Phase III clinical trials. The results of the first trials were disappointing, with a failure to meet the study goals,[2][3][4] Additional Phase III trials are ongoing.[2]
Dacomitinib is a HER (erbB) inhibitor in clinical trial development at Pfizer for the treatment of advanced non-small cell lung cancer (NSCLC) and for the treatment of relapsed/recurrent glioblastoma.
No recent development has been reported for research into the treatment of recurrent and/or metastatic head and neck squamous cell cancer. In 2012, Pfizer and SFJ Pharmaceuticals signed a codevelopment agreement for dacomitinib for the treatment of patients with locally advanced or metastatic NSCLC with activating mutations of epidermal growth factor receptor.
No recent development has been reported for research into the treatment of recurrent and/or metastatic head and neck squamous cell cancer. In 2012, Pfizer and SFJ Pharmaceuticals signed a codevelopment agreement for dacomitinib for the treatment of patients with locally advanced or metastatic NSCLC with activating mutations of epidermal growth factor receptor.
Substituted 4-phenylamino-quinazolin-6-yl-amides useful in the treatment of cancer have been described in the art, including those of U.S. Pat. No. 5,457,105 (Barker), U.S. Pat. No. 5,760,041 (Wissner et al.), U.S. Pat. No. 5,770,599 (Gibson), U.S. Pat. No. 5,929,080 (Frost), U.S. Pat. No. 5,955,464 (Barker), U.S. Pat. No. 6,251,912 (Wissner et al.), U.S. Pat. No. 6,344,455 (Bridges et al.), U.S. Pat. No. 6,344,459 (Bridges et al.), U.S. Pat. No. 6,414,148 (Thomas et al.), U.S. Pat. No. 5,770,599 (Gibson et al.), U.S. patent application 2002/0173509 (Himmelsbach et al.), and U.S. Pat. No. 6,323,209 (Frost).
Dacomitinib is a pan-human epidermal growth factor receptor (pan-HER) inhibitor developed by Pfizer, as ー small molecules targeting ffiR-1, HER-2 and HER-4 tyrosine kinase inhibitor by irreversibly binding to HER-l, HER-2, HER-4 and anti-tumor effect. Ni-line treatment of non-small cell lung cancer (NSCLC) display, Dacomitinib in non-small cell lung cancer Dinner erlotinib compared to some extend on progression-free survival and quality of life have mentioned the smell.
_4] Structural formula for Dacomitinib

[0005] U.S. patent US7772243 Dacomitinib first proposed a synthesis method, first, a fluorine-2_ _4_ amino acid and formamidine ring closure reaction to give 7 – fluoro-4 – quinazolinone, nitration and then successively chlorination reaction, to give 4 – chloro-7 – fluoro-6 – nitro-quinazoline; another aspect ー 3 – chloro-4 – amino-substituted on a fluoroaniline to give 3 – chloro – # – (3,4 – ni section yl methoxy)-4_ fluoro-aniline, obtained after the coupling of both an amino-protected N-(3 – chloro-4 – fluorophenyl)-7 – fluoro-6 – nitro-quinazoline -4 – amine, protected amino N-(3 – chloro-4 – fluorophenyl
Yl)-7_ fluoro-6 – nitro-quinazolin-4 – amine is of formula

Followed by a methoxy group, an amidation reaction and hydrogenation, the final deprotection ko under the action of trifluoroacetic acid to give the final product Dacomitinib. Throughout the reaction as follows:

synthesis
Synthesis ー kind EGFR inhibitors Dacomitinib, synthetic route for

A synthetic method EGFR inhibitors Dacomitinib, concrete steps are as follows:
Step I, 7 – fluoro-4 – Synthesis of quinazolinone:

30 g (0.1934mol) 2 – fluoro-amino acid was dissolved in 250 ml _4_ formamide among the reaction was heated to 150 ° C for 6 inch, TLC plates to determine the point of completion of the reaction. The reaction was poured hot into 2000 ml of ice water, filtered, the filter cake was washed with water, vacuum dried at 50 ° C for 14 hours to give a pale brown solid powder 7 – fluoro-4 – quinazolinone, 28 g, yield 88%.
[0021] 2 walk 7 – fluoro-6 – nitro-4_ (hydrogen) _ Synthetic quinazolinones of:

Concentrated sulfuric acid (50 ml) and fuming nitric acid (50 ml) mixture was cooled with an ice bath to (TC hereinafter under stirring slowly added 25 g (0.1523mol) 7 – fluoro-4 – quinazolinone , the addition was complete, the reaction mixture was stirred at room temperature for I hour and then the reaction was heated to 110 ° C for 2 inch, TLC plates to determine the point of completion of the reaction the reaction was cooled to room temperature, 300 ml of ice water, the precipitated solid was stirred for 30 minutes , filtered, the filter cake was washed with water, vacuum dried at 50 ° C in 14 hours to give a yellow solid powder 7 – fluoro-6 – nitro-4 – (hydrogen) – quinazolinone, 26 g, yield 82%.
[0022] Step 3 6 – amino-7 – fluoro-4 – (hydrogen) – quinazolinone Synthesis:

24 g (0.1148mol) 7 – fluoro-6 – nitro _4_ (hydrogen) – quinazolinone was dissolved in 400 ml of methanol was added 2 g of palladium / carbon catalyst was added 8 ml of concentrated hydrochloric acid, and hydrogen was 2 small inch atmospheric reaction, TLC plates to determine the point of the reaction is complete. The catalyst was removed by suction filtration through celite, washed several fitness methanol, and the filtrate was concentrated by rotary evaporation to dryness to give 6 – amino-7_ fluoro-4 – (hydrogen) – quinazolinone, yellow powder, 20 g, yield 97%.
[0023] 4 walk, ⑶ -4 – (piperidin – Suites yl) -2 – butene acid methyl ester synthesis:
18 g (0.1006mol) 4 – bromo-methyl crotonate dissolved in 180 ml of methylene chloride ni added 27.9 g (0.2019mol) potassium carbonate, cooled to ice-bath (TC, was slowly added dropwise 10 ml (0.1012mol ) piperidine, (I reaction was stirred under a small inch TC, TLC plates to determine the point of completion of the reaction was concentrated by rotary evaporation to dryness, to give (E) -4 – (piperidin-1 – yl) – 2 – butenoic acid methyl Cool as a yellow solid, 17.1 g, yield 93%.
[0024] 5th walk, Buddhist) -4 – (piperidin-1 – yl) -2 – butene acid hydrochloride synthesis:
16 g (0.0873mol) of W) -4 – (piperidin-_1_ yl) -2 – butenyl acetate and 80 ml of concentrated hydrochloric acid was added to 250 ml of 1,4 – ni oxygen dioxane, heated under reflux 20 hours inch, TLC plate point the reaction was determined complete, the reaction solution was concentrated by rotary evaporation to dryness surplus was recrystallized from isopropanol to give a pale yellow solid, Buddhist) _4-(piperidin-1 – yl) -2 – butene acid hydrochloride, 14.5 g, yield 81%.
[0025] Step 6, (E) -4 – (piperazine Jie fixed -1 – yl) – 2 – butenyl chloride synthesis:
13 g (0.0632mol) of (K) ~ 4 ~ (piperidin-1 – yl) -2 – butene acid hydrochloride was dissolved in 750 ml of methylene chloride ni, 5 ml of DMF, was slowly added dropwise 8 ml ( 0.0933mol) of oxalyl chloride, the reaction was stirred at room temperature for I h, TLC plates to determine the point of completion of the reaction, the reaction solution was concentrated to dryness by rotary evaporation to give a pale yellow oil, Buddhist) _4-(piperidin-1 – yl) -2 – butyl allyl chloride, 11.8 g, yield 99%.
[0026] Step 7 (cargo) – # – (7 – fluoro-4 – oxo-3 ,4 – ni hydrogen quinazolin-6 – yl) -4 – (piperidin-1 – yl) -2 – butene amide Synthesis:

11 g (0.0586mol) of the) -4 – (piperidin-1 – yl) – 2 – butenyl chloride ni chloride (50 ml) was slowly added dropwise to 6 – amino-1 – fluoro-4 – ( hydrogen) – quinazolinone (7 g, 0.0391mmol), three ko amine (14 ml) and the mixture was ni chloride (200 ml), the reaction mixture was stirred at room temperature for 2 hours the reaction inch, TLC determined the completion of reaction points board , was added 800 liters of halo ni halo chloroformate and 500 liters of burning the separated organic phase was washed with 500 liters of halo, halo and then with 500 liters of brine, dried over magnesium sulfate, and concentrated by rotary evaporation to dryness was subjected to silica gel surplus Column chromatography (30% acid ko ko acetate / hexane) to give (M)-N-(7 – fluoro-4 – oxo-3 ,4 – ni hydrogen quinazolin-6 – yl) -4 – (piperidin-1 – yl) -2 – butenamide, as a pale yellow solid, 12.3 g, yield 95%.
Step 8 [0027] (2 ^) – # – (7 – methoxy – 4 – oxo _3, 4_ ni hydrogen quinazolinyl _6_ yl)-4_ (piperidin-1 – yl) – Synthesis 2_ butenamide:
I ^ xN MeONa N. Under nitrogen atmosphere, to 100 ml of anhydrous methanol was slowly added 1.52 g of sodium metal (0.0661mol), stirred for 10 minutes to dissolve all of the sodium metal to the completion of the reaction, to obtain a freshly prepared solution of sodium methoxide, and the The sodium methoxide solution was added 11 g (0.0333mol) of (receive) (7 – fluoro-4 – oxo-3 ,4 – ni hydrogen quinazolin-6 – yl) -4 – (piperidin-1 – yl) 2_ butene-amide, the reaction was heated to reflux for 3 inch, TLC plates to determine completion of the reaction point, cooled to room temperature, acidified with 2N hydrochloric acid solution to pH = 3 ~ 4, and concentrated by rotary evaporation to dryness, the residue was washed with water beating, filtration, The filter cake was washed with water, vacuum dried at 50 ° C in 14 hours to give (article) – # – (7 – methoxy – 4 – oxo – ni hydrogen quinazolin-6 – yl) – 4_ (piperidin-1 – yl)-2_ butenamide yellow solid, 10.6 g, yield 93%.
[0028] Step 9, {W,-N-(4 – chloro-7 – methoxy-quinazoline _6_ yl)-4_ (piperidin _1_ yl)-amide <EMI butene 2_:
9 g (0.0263mol) of (receive) – # – (7 – methoxy _4_ oxo – ni hydrogen quinazolin-6 – yl)-4_ (piperidin-1 – yl) – 2_ butenamide were added to 40 ml of phosphorus oxychloride was heated under reflux for 2 inch, TLC plates to determine the point of completion of the reaction, the reaction solution was concentrated to dryness by rotary evaporation, ice water was added surplus, beating, filtered, the cake washed with washed with water, vacuum dried at 50 ° C in 14 hours to give {W,-N-(4 – chloro-7 – methoxy-quinazolin-6 – yl) -4 – (piperidin-1 – yl) – 2 – butene amide as a yellow solid, 7 g, yield 74%

(2E)-N-(4 – chloro-7 – methoxy-quinazolin-6 – yl) -4 – (piperidin-1 – yl) -2 – butene amide (6 g,
0.0166mol), 3 – chloro-4-fluoro-aniline (2.6 g, 0.0179mol) and three ko amine (2.6 ml, 0.0186mol) was added to 140 ml of isopropanol and the reaction was heated to reflux for 3 inch, TLC plates to determine the point completion of the reaction, cooled to room temperature, filtered, the filter cake washed with methanol, vacuum dried at 50 ° C in 14 hours to give the final product Dacomitinib, a yellow solid, 6.6 g, yield 84%.
/////////////////////////
synthesis
US7772243
Scheme 1, wherein the 4-position aniline group is represented a 4-fluoro-3-chloro aniline group.

4-Chloro-7-fluoro-6-nitroquinazoline (7) can be prepared by methods similar to those described in J.Med. Chem. 1996, 39, 918-928. Generally, 2-amino-4-fluoro-benzoic acid (1) can be reacted with formamidine (2) and acetic acid (3) in the presence of 2-methoxyethanol to provide 7-Fluoro-3H-quinazolin-4-one (4). The 7-fluoro-3H-quinazolin-4-one (4) can be nitrated to 7-fluoro-6-nitro-3H-quinazolin-4-one (5), which can be treated with thionyl chloride to yield 4-chloro-6-nitro-7-fluoro-3H-quinazoline (6). The 4-chloro-quinazoline compound (6) can be combined with a desirably substituted aniline, represented above by 4-fluoro-3-chloro-aniline, in the presence of a tertiary amine and isopropanol to provide the 4-anilino-6-nitro-7-fluoro-quinazoline (7).
The 4-anilino-6-nitro-7-fluoro-quinazoline (7) may be reacted with an alcohol of the formula R3OH, wherein R3 is as defined above, to yield the 7-alkoxylated compound (8). Reduction of the 6-nitro compound (8) provides the 6-amino analog (9).
The 6-position amino compound (9) may be reacted with a haloalkenoyl chloride (12), such as a 4-bromo-but-2-enoyl chloride, 5-bromo-pent-2-enoyl chloride, 4-chloro-but-2-enoyl chloride, or 5-chloro-pent-2-enoyl chloride, to provide an alkenoic acid[4-anilino]-7-alkoxylated-quinazolin-6-yl-amide (13). Haloalkenoyl chloride agents useful in this scheme may be prepared by methods known in the art, such as the treatment of a relevant haloalkenoic acid, represented by bromoalkenoic acid ester (10), with a primary alcohol, yielding the corresponding haloalkenoic acid (11), which may in turn be treated with oxalyl chloride to provide the desired haloalkenoyl chloride (12).
Finally, the quinazoline-6-alkanoic acid compound (13) may be treated with a cyclic amine, such as piperidine, piperazine, etc., to provide the desired final compound (14).
EXAMPLE 2
4-Piperidin-1-yl-but-2-enoic acid [4-(3-chloro-4-fluoro-phenylamino)-7-methoxy-quinazolin-6-yl]-amide (Synthetic Route No. 1)

The title compound and other 7-methoxy analogs of this invention can be prepared as described in Example 1 by replacing the 2-fluoroethanol used in Example 1 with stoichiometric amount of methanol.
EXAMPLE 3 4-Piperidin-1 -yl-but-2-enoic acid [4-(3-chloro-4-fluoro-phenylamino)-7-methoxv -quinazolin-6-yl]-amide (Synthetic Route No. 2)
An alternative synthetic route for compounds of this invention involves preparing the 6-position substituent chain as a Het-alkenoyl chloride as depicted in Scheme 2, below.


It will be understood that other compounds within this invention may be prepared using Het-butenoyl halide, Het-pentenoyl halide and Het-hexenoyl halide groups of the formula:
wherein R4 is as described herein and halo represents F, Cl, Br or I, preferably Cl or Br. One specific group of these Het-alkenoyl halides includes those compounds in which halo is Cl or Br, R4 is —(CH2)m-Het, m is an integer from 1 to 3, and Het is piperidine or the substituted piperidine moieties disclosed above.
EXAMPLE 4 4-Piperidin-1-yl-but-2-enoic acid [4-(3-chloro-4-fluoro-phenylamino)-7-methoxy-quinazolin-6-yl]-amide (Synthetic Route No. 3)


3-Chloro-4-fluoro-phenylamine 15 (50.31, 345.6 mmole) and 3,4-Dimethoxy-benzaldehyde 16 (57.43 g, 345.6 mmole) were mixed in 500 ml of IPA and cooled in an ice-water. The glacial acetic acid was added (20.76 g, 345.6 mole) and then sodium cyanoborohydride in one portion. The reaction was stirred at room temperature (RT) for 24 hrs. 250 mL of 10% NaOH was added dropwise at RT after the reaction was completed. The mixture was stirred for ½ hr. The slurry was then filtered and washed with IPA and dried in vacuo. The mass weight 88.75 g (17, 87%).
Compounds 6 (3 g, 13.18 mmole) and 17 (3.9 g, 13.18 mmole) were combined in CH3CN (25 mL) and heated for one hr. Mass spectroscopy indicated no starting material. Saturated K2CO3 was added and the reaction was extracted 3× with EtOAc. The organic layers were combined, washed with brine and concentrated in vacuo to give 6.48 g of 7 (78.4%).
Compound 7 (72.76 g, 149.4 mmole) was added to a cool solution of NaOMe in 1.5 L of dry MeOH under N2. The cooling bath was removed and the mixture was heated to reflux and stirred for 1 hr. The reaction was cooled to room temperature and quenched with water until the product precipitated out. The solid was filtered and washed with water and hexanes. The product was slurred in refluxing EtOAc and filtered hot to provide 68.75 g of yellow soled 8 (73%).
Compound 8 (63.62 g, 127.5 mole) was hydrogenated using Raney/Ni as catalyst to obtain 43.82 g of 9 (100%). Oxalyl chloride (6.5 g, 51.18 mmole) was added slowly to a suspension of 13 (10.5 g, 51.2 mmole) in 200 ml of dichloromethane containing 8 drops of DMF, after the reaction become homogeneous, the solvent was removed and the residual light yellow solid was slurred in 200 ml of DMAC and 9 (20 g, 42.65 mmole) was added gradually as a solid. The reaction was stirred for 15 min. and poured slowly into 1N NaOH. The mixture was extrated 3× EtOAc. The combined organic layers were washed with brine, filtered and concentrated in vacuo to obtain 28.4 g (100%) 10.
Compound 10(13.07 g, 21.08 mmole) was dissolved in trifluoroacetic acid (TFA) (74 g, 649 mmole) and heated to 30° C. for 24 hrs. The reaction was cooled to RT and poured gradually into a cooled 1 N NaO H-brine solution. Precipitate formed and was filtered and washed with 3X water then dried. The precipitate was recrystallized from toluene to obtain pure 4-Piperidin-1-vl-but-2-enoic acid [4-(3-chloro-4-fluoro-phenylamino) -7-methoxv-puinazolin-6-yl]-amide (9.90 g, 89%).
Example 1 is similar but not same…caution
EXAMPLE 1 4-Piperidin-1-yl-but-2-enoic acid [4-(3-chloro-4-fluoro-phenylamino)-7-(2-fluoro-ethoxy)-quinazolin-6-yl]-amide

7-fluoro-6-nitro-4-chloroquinazoline (14.73,g, 65 mmol) was combined with 3-choro-4-fluoroaniline (9.49 g, 65 mmol) and triethylamine (10 mL, 72 mmol) in 150 mL of isopropanol. The reaction was stirred at room temperature for 1.5 hours, resulting in a yellow slurry. The solid was collected by filtration, rinsing with isopropanol and then water. The solid was dried in a 40° C. vacuum oven overnight to give 19.83 g (91%) of the product as an orange solid.
MS (APCI, m/z, M+1): 337.0
NaH (60% in mineral oil, 3.55 g, 88 mmol) was added, in portions, to a solution of 2-fluoroethanol (5.19 g, 80 mmol) in 200 mL THF. The reaction was stirred for 60 minutes at room temperature. To the reaction was added 7-fluoro-6-nitro-4-(3-chloro-4-fluoroaniline)quinazoline (18.11 g, 54 mmol) as a solid, rinsing with THF. The reaction was heated to 65° C. for 26 hours. The reaction was cooled to room temperature and quenched with water. THF was removed in vacuo. The resulting residue was sonicated briefly in water then the solid collected by filtration. The solid was triturated with MeOH, filtered and dried in a 40° C. vacuum oven overnight to 12.63 g of the product. Additional product was obtained by concentrating the MeOH filtrate to dryness and chromatography eluting with 50% EtOAc/hex. The isolated material was triturated with MeOH (2×), filtered and dried. 3.90 g
Total yield: 16.53 g, 81%
MS (APCI, m/z, M+1): 381.0
7-(2-fluoroethoxy)-6-nitro-4-(3-chloro-4-fluoroaniline)quinazoline (0.845 g, 2.2 mmol) in 50 mL THF was hydrogenated with Raney nickel (0.5 g) as the catalyst over 15 hours. The catalyst was filtered off and the filtrate was evaporated to give 0.77 g of product. (99%)
MS (APCI, m/z, M+1): 351.2
Methyl 4-bromocrotonate (85%, 20 mL, 144 mmol) was hydrolyzed with Ba(OH)2 in EtOH/H2O as described in J.Med.Chem. 2001, 44(17), 2729-2734.
MS (APCI, m/z, M−1): 163.0
To a solution of 4-bromocrotonic acid (4.17 g, 25 mmol) in CH2Cl2 (20 mL) was added oxalyl chloride (33 mL, 38 mmoL) and several drops of DMF. The reaction was stirred at room temperature for 1.5 hours. The solvent and excess reagent was removed in vacuo. The resulting residue was dissolved in 10 mL THF and added to a 0° C. mixture of 6-amino-7-(2-fluoroethoxy)-4-(3-chloro-4-fluoroaniline)quinazoline (5.28 g, 15 mmol) and triethylamine (5.2 mL, 37 mmol). The reaction was stirred at 0° C. for 1 hour. Water was added to the reaction and the THF removed in vacuo. The product was extracted into CH2Cl2 (400 mL). The organic layer was dried over MgSO4, filtered and concentrated. The crude material was chromatographed on silica gel eluting with 0-4% MeOH/CH2Cl2. An isolated gold foam was isolated. Yield: 4.58 g, 61%
MS (APCI, m/z, M−1): 497.1
Piperidine (0.75 mL, 6.7 mmol) was added to a solution of the above compound (3.35 g, 6.7 mmol) and TEA (2.80 mL, 20 mmol) in 10 mL DMA at 0° C. The reaction was stirred at 0° C. for 17 hours. Water was added to the reaction until a precipitate was evident. The reaction was sonicated for 40 minutes and the liquid decanted. The residue was dissolved in CH2Cl2, dried over MgSO4, filtered and concentrated. The material was chromatographed on silica gel eluting with 4-10% MeOH/CH2Cl2. The isolated residue was triturated with acetonitrile (2×) and collected by filtration. Impurity found: Michael addition of piperidine (2.2% in first trituration of acetonitrile). Additional material can be obtained from the acetonitrile filtrates.
Yield: 0.95 g, 27%
MS (APCI, m/z, M+1): 502.3
……………
US 20050250761 A1,
References
- “Dacomitinib”. NCI Drug Dictionary.
- Zosia Chustecka (January 27, 2014). “Dacomitinib Fails in Pretreated Non-small Cell Lung Cancer”. Medscape.
- “Blow to Pfizer as dacomitinib fails in lung cancer trials”. pmlive.com. 28th January 2014.
- “Pfizer Announces Top-Line Results From Two Phase 3 Trials Of Dacomitinib In Patients With Refractory Advanced Non-Small Cell Lung Cancer”. Pfizer Press Release. January 27, 2014.
- Tyrosine kinase inhibitors.17. Irreversible inhibitors of the epidermal growth factor receptor: 4-(Phenylamino)quinazoline- and 4-(phenylamino)pyrido[3,2-d]pyrimidine-6-acrylamides baring additional solubilizing functions
J Med Chem 2000, 43(7): 1380
- Dacomitinib Fact Sheet, Pfizer
| WO1996033980A1 * | 23 Apr 1996 | 31 Oct 1996 | Keith Hopkinson Gibson | Quinazoline derivatives |
| WO1997038983A1 * | 8 Apr 1997 | 23 Oct 1997 | Alexander James Bridges | Irreversible inhibitors of tyrosine kinases |
| WO2002050043A1 * | 12 Dec 2001 | 27 Jun 2002 | Boehringer Ingelheim Pharma | Quinazoline derivatives, medicaments containing said compounds, their utilization and method for the production thereof |
| WO2004069791A2 * | 3 Feb 2004 | 19 Aug 2004 | Hubert Gangolf Klemens Barth | Preparation of substituted quinazolines |
| US5760041 * | 21 Jan 1997 | 2 Jun 1998 | American Cyanamid Company | 4-aminoquinazoline EGFR Inhibitors |
12
MOMELOTINIB

Momelotinib
414.47, C23H22N6O2,
1056634-68-4
N-(Cyanomethyl)-4-[2-(4-morpholin-4-ylanilino)pyrimidin-4-yl]benzamide
N-(Cyanomethyl)-4-[2-[4-(4-morpholinyl)phenylamino]pyrimidin-4-yl]benzamide
Jak2 tyrosine kinase inhibitor; Jak1 tyrosine kinase inhibitor
Inflammatory disease; Myelofibrosis; Myeloproliferative disorder; Pancreatic ductal adenocarcinoma; Polycythemia vera
CYT 387; CYT-387; momelotinib)
GS-0387


CYT387 is an ATP-competitive small molecule JAK1 / JAK2 inhibitor with IC50 of 11 and 18 nM for JAK1 and JAK2, respectively. CYT387 is useful for treatment of myeloproliferative disorders and anti-cancer.
CYT-387 is a potent, orally administered JAK1/JAK2/ Tyk2 inhibitor in phase III clinical studiest at Gilead for the treatment of post-polycythemia vera, for the treatment of primary myelofibrosis and for the treatment of post-essential thrombocythemia. Phase II studies are also ongoing, in combination with gemcitabine and nab-paclitaxel, in adults with untreated metastatic pancreatic ductal adenocarcinoma.
The compound possesses an excellent selectivity and safety profile. In 2010 and 2011, orphan drug designation was assigned by the FDA and the EMA, respectively, for the treatment of myelofibrosis. In 2011, orphan drug designation was assigned by the EMA for the treatment of post-essential thrombocythemia myelofibrosis and for the treatment of post-polycythemia vera myelofibrosis.
………………….
N-(cyanomethyl)-4-(2-(4-morpholinophenylamino)pyrimidin-4-yl)benzamide
| 3 |  | 414.18 | 1H NMR (300 MHz, d6-DMSO): δ 9.47 (1 H, s), 9.32 (1 H, t, J = 5.5 Hz), 8.54 (1 H, d, J = 5.0 Hz), 8.27 (2 H, d, J = 8.7 Hz), 8.02 (2 H, d, J = 8.2 Hz), 7.67 (2 H, d, J = 9.1 Hz), 7.41 (1 H, d, J = 5.5 Hz), 6.93 (2 H, d, J = 9.1 Hz), 4.36 (2 H, d, J = 5.5 Hz), 3.75 (4 H, m), 3.05 (4 H, m). | m/z 415.3 [M + H]+ | N-(cyanomethyl)-4-(2-(4- morpholinophenylamino)pyrimidin- 4-yl)benzamide |
Example 1Synthesis of Compound 3
A mixture of 4-ethoxycarbonylphenyl boronic acid (23.11 g, 119 mmol), 2,4-dichloropyrimidine (16.90 g, 113 mmol), toluene (230 mL) and aqueous sodium carbonate (2 M, 56 mL) was stirred vigorously and nitrogen was bubbled through the suspension for 15 minutes. Tetrakis(triphenylphosphine)palladium[0] (2.61 g, 2.26 mmol) was added. Nitrogen was bubbled through for another 10 min., the mixture was heated to 100° C., then at 75° C. overnight. The mixture was cooled, diluted with ethyl acetate (200 mL), water (100 mL) was added and the layers were separated. The aqueous layer was extracted with ethyl acetate (100 ml) and the two organic extracts were combined. The organics were washed with brine, filtered through sodium sulfate, concentrated, and the resultant solid was triturated with methanol (100 mL) and filtered. The solids were washed with methanol (2×30 mL) and air dried. This material was dissolved in acetonitrile (150 mL) and dichloromethane (200 mL), stirred with MP.TMT Pd-scavenging resin (Agronaut part number 800471) (7.5 g) over 2 days. The solution was filtered, the solids were washed with dichloromethane (2×100 mL), and the filtrate concentrated to give ethyl 4-(2-chloropyrimidin-4-yl)benzoate as an off-white solid (17.73 g, 60%)—additional washing with dichloromethane yielded a further 1.38 g and 0.5 g of product. 1H NMR (300 MHz, d6-DMSO) δ 8.89 (1H, d, J=5.0 Hz); 8.32 (2H, d, J=8.7 Hz); 8.22 (1H, d, J=5.5 Hz); 8.12 (2H, d, J=8.7 Hz); 4.35 (2H, q, J=7.1 Hz); 1.34 (3H, t, J=7.1 Hz); LC-ESI-MS (method B): rt 7.3 min.; m/z 263.0/265.0 [M+H]+.
A mixture of ethyl 4-(2-chloropyrimidin-4-yl)benzoate (26.15 g, 99.7 mmol) and 4-morpholinoaniline (23.10 g, 129.6 mmol) was suspended in 1,4-dioxane (250 mL). p-Toluenesulfonic acid monohydrate (17.07 g, 89.73 mmol) was added. The mixture was heated at reflux for 40 h., cooled to ambient temperature, concentrated then the residue was partitioned between ethyl acetate and 1:1 saturated sodium bicarbonate/water (1 L total). The organic phase was washed with water (2×100 mL) and concentrated. The aqueous phase was extracted with dichloromethane (3×200 mL). The material which precipitated during this workup was collected by filtration and set aside. The liquid organics were combined, concentrated, triturated with methanol (200 mL) and filtered to yield additional yellow solid. The solids were combined, suspended in methanol (500 mL), allowed to stand overnight then sonicated and filtered. The solids were washed with methanol (2×50 mL) to give, after drying, ethyl 4-(2-(4-morphonlinophenylamino)pyrimidin-4-yl)benzoate (35.39 g, 88%). 1H NMR (300 MHz, d6-DMSO) δ 9.49 (1H, s); 8.54 (1H, d, J=5.0 Hz); 8.27 (2H, d, J=8.7 Hz); 8.10 (2H, d, J=8.7 Hz), 7.66 (2H, d, J=9.1 Hz); 7.38 (1H, d, J=5.0 Hz); 6.93 (2H, d, J=8.7 Hz); 4.35 (2H, q, J=6.9 Hz), 3.73 (4H, m); 3.04 (4H, m); 1.34 (3H, t, J=6.9 Hz); LC-ESI-MS (method B): rt 7.5 min.; m/z 404.1 [M+H].
A solution of ethyl 4-(2-(4-morpholinophenylamino)pyrimidin-4-yl)benzoate (35.39 g, 87.6 mmol) in 3:1 methanol/tetrahydrofuran (350 mL) was treated with lithium hydroxide (4.41 g, 183.9 mmol) in water (90 mL). The mixture was heated at reflux for 2 h., cooled, concentrated and acidified with hydrochloric acid (2M, 92.5 mL, 185 mmol). The dark precipitate was filtered, washed with water, and dried under vacuum. The solid was ground to a powder with a mortar and pestle, triturated with methanol (500 mL) then filtered again to yield 4-(2-(4-morpholinophenylamino)pyrimidin-4-yl)benzoic acid as a muddy solid. This material was washed with ether, air dried overnight, and ground to a fine powder with mortar and pestle. On the basis of mass recovery (34.49 g) the yield was assumed to be quantitative. 1H NMR (300 MHz, d6-DMSO) δ 9.47 (1H, s); 8.53 (1H, d, J=5.2 Hz); 8.24 (2H, d, J=8.5 Hz); 8.08 (2H, d, J=8.8 Hz), 7.66 (2H, d, J=9.1 Hz); 7.37 (1H, d, J=5.2 Hz); 6.93 (2H, d, J=9.1 Hz); 3.73 (4H, m); 3.04 (4H, m). LC-ESI-MS (method C): rt 7.3 min.; m/z 377.1 [M+H]+.
To a suspension of 4-(2-(4-morpholinophenylamino)pyrimidin-4-yl)benzoic acid (theoretically 32.59 g, 86.6 mmol) in DMF (400 mL) was added triethylamine (72.4 mL, 519.6 mmol, 6 eq.) The mixture was sonicated to ensure dissolution. Aminoacetonitrile hydrochloride (16.02 g, 173.2 mmol) was added followed by N-hydroxybenzotriazole (anhydrous, 14.04 g, 103.8 mmol) and 1-ethyl-3-(dimethylaminopropyl)carbodiimide hydrochloride (19.92 g, 103.8 mmol). The suspension was stirred vigorously overnight. The solvent was evaporated under reduced pressure, the residue was diluted with 5% sodium bicarbonate (400 mL) and water (300 mL), giving a yellow solid, which was broken up and filtered. The solids were washed several times with 100 mL portions of water, triturated with hot methanol/dichloromethane (500 mL, 1:1), concentrated to a volume of approximately 300 mL), cooled and filtered. The solids were washed with cold methanol (3×100 mL), ether (200 mL) and hexane (200 mL) prior to drying to afford
Compound 3 (31.69 g, 88%). M.p. 238-243° C.
Microanalysis: Found C, 66.52; H, 5.41; N, 20.21. C23H26N6O10S2 requires C, 66.65; H, 5.35; N, 20.28%.
13C NMR (75.5 MHz, d6-DMSO) δ 166.04, 162.34, 160.26, 159.14, 146.14, 139.87, 134.44, 132.73, 127.80, 126.84, 120.29, 117.49, 115.50, 107.51, 66.06, 49.16, 27.68.
1H NMR GIVEN ABOVE
Example 6Salt Formation from Compound 3
Compound 3 (10.0 g) was suspended in methanol (1 L). Concentrated sulfuric acid (10.52 g, 90% w/w) was added dropwise to the stirring solution. A clear brown solution resulted and a solid lump formed. The solution was filtered quickly then allowed to continue stirring for 3 h (a second precipitate appeared within minutes). After this time the pale yellow precipitate was collected by filtration, washed with methanol (10 mL) then dried under vacuum overnight to afford 4-(4-(4-(4-(cyanomethylcarbamoyl)phenyl)pyrimidin-1-ium-2-ylamino)phenyl)morpholin-4-ium hydrogensulfate, as a pale yellow solid (10.20 g, 69%). m.p. 205° C. Microanalysis: Found C, 45.18; H, 4.36; N, 13.84; S, 10.24. C23H26N6O10S2 requires C, 45.24; H, 4.29; N, 13.76; S 10.50%. 1H NMR (300 MHz, d6-DMSO) δ 9.85 (br. s, 1H), 9.34 (t, J=5.4 Hz, 1H), 8.59 (d, J=5.2 Hz, 1H), 8.27 (d, J=8.5 Hz, 2H), 8.03 (d, J=8.5 Hz, 2H), 7.83 (d, J=8.4 Hz, 2H), 7.50 (d, J=5.2 Hz, 1H), 7.34 (br. s, 2H), 4.36 (d, J=5.4 Hz, 2H), 3.89 (br. s, 4H), 3.37 (br. s, 4H); 13C NMR (75.5 MHz, d6-DMSO) δ 166.07, 163.36, 159.20, 158.48, 140.19, 139.34, 136.45, 134.89, 128.00, 127.22, 121.13, 119.89, 117.59, 109.05, 64.02, 54.04, 27.82. LC-ESI-MS (method D): rt 10.0 min.; m/z 415.1 [M+H]+.
Compound 3 (0.25 g) was suspended in methanol (25 ml). Methane sulfonic acid (0.255 g) was added dropwise to the stirring solution and a clear brown solution resulted. The solution was allowed to stir for 3 h, after which the volume was reduced to 9 ml. The resultant precipitate was collected and dried under vacuum for 8 h to afford 4-(4-(4-(4-(cyanomethylcarbamoyl)phenyl)pyrimidin-1-ium-2-ylamino)phenyl)morpholin-4-ium methanesulfonate as a pale yellow solid (0.22 g). m.p. 208° C. 1H NMR (300 MHz, d6-DMSO) δ 9.83 (br. s, 1H), 9.35 (t, J=5.3 Hz, 1H), 8.59 (d, J=5.1 Hz, 1H), 8.28 (d, J=8.5 Hz, 2H), 8.04 (d, J=8.5 Hz, 2H), 7.83 (d, J=9.0 Hz, 2H), 7.50 (d, J=5.5 Hz, 1H), 7.31 (d, J=9.0 Hz, 2H), 4.36 (d, J=5.5 Hz, 2H), 3.88 (m, 4H), 3.35 (br. s, 4H), 2.36 (s, 6H); LC-ESI-MS (method D): rt 10.2 min.; m/z 415.3 [M+H]+.
Compound 3 (0.50 g) was suspended in methanol (45 ml). A freshly prepared solution of hydrochloric acid in methanol (2.6 ml, HCl conc. 40 mg/ml) was added dropwise to the stirring solution and a clear brown solution resulted. The solution was allowed to stir for 2 h, then the resultant precipitate was collected, washed with methanol (5 ml) and dried under vacuum for 8 h to afford 4-(4-(4-(4-(cyanomethylcarbamoyl)phenyl)pyrimidin-1-ium-2-ylamino)phenyl)morpholin-4-ium chloride a pale yellow solid (0.30 g). m.p. 210° C.1H NMR (300 MHz, d6-DMSO) 1H NMR (300 MHz, DMSO) δ 9.92 (br. s, 1H), 9.42 (t, J=5.3, 1H), 8.62 (d, J=4.8, 1H), 8.29 (d, J=8.1, 2H), 8.06 (d, J=8.1, 2H), 7.89 (d, J=9.0, 2H), 7.53 (br. s, 3H), 4.36 (d, J=5.4, 2H), 3.82 (br. s, 4H), 3.43 (br. s, 4H)
LC-ESI-MS (method D): rt 10.3 min.; m/z 415.3 [M+H]+.
………………………………….
SEE
WO 2014114274
References on CYT387
. [1] A Pardanani et al CYT387, a Selective JAK1 / JAK2 inhibitor: in vitroassessment of kinase selectivity and preclinical s using Cell lines and Primary cells from polycythemia vera Patients Leukemia (2009) 23, 1441-1445
Abstract
Somatic mutations in Janus kinase 2 (JAK2), including JAK2V617F, result in dysregulated JAK-signal transducer and activator transcription (STAT) signaling, which is implicated in myeloproliferative neoplasm (MPN) pathogenesis. CYT387 is an ATP-competitive small molecule that potently inhibits JAK1 / JAK2 kinases ( IC (50) = 11 and 18 nM, respectively), with significantly less activity against other kinases, including JAK3 (IC (50) = 155 nM). CYT387 inhibits growth of Ba / F3-JAK2V617F and human erythroleukemia (HEL) cells ( IC (50) approximately 1500 nM) or Ba / F3-MPLW515L cells (IC (50) = 200 nM), but has considerably less activity against BCR-ABL harboring K562 cells (IC = 58 000 nM). Cell lines harboring mutated JAK2 alleles (CHRF-288-11 or Ba / F3-TEL-JAK2) were inhibited more potently than the corresponding pair harboring mutated JAK3 alleles (CMK or Ba / F3-TEL-JAK3), and STAT-5 phosphorylation was inhibited in HEL cells with an IC (50) = 400 nM. …
[2]. Tyner Jeffrey W. et al CYT387, a novel JAK2 inhibitor, induces Hematologic Responses and normalizes inflammatory cytokines in murine myeloproliferative neoplasms Blood June 24, 2010vol. no 115. 255232-5240
Abstract
Activating alleles of Janus kinase 2 (JAK2) SUCH as JAK2 (V617F) are Central to the pathogenesis of myeloproliferative neoplasms (MPN), suggesting Small molecule inhibitors targeting JAK2 That May be therapeutically Useful. IDENTIFIED We have an aminopyrimidine derivative ( CYT387), which inhibits JAK1, JAK2, and tyrosine kinase 2 (TYK2) at low nanomolar concentrations, with few additional targets. Between 0.5 and 1.5muM CYT387 caused growth suppression and apoptosis in JAK2-dependent hematopoietic cell lines, while nonhematopoietic cell lines were unaffected. In a murine MPN model, CYT387 normalized white cell counts, hematocrit, spleen size, and restored physiologic levels of inflammatory cytokines. Despite the hematologic responses and reduction of the JAK2 (V617F) allele burden, JAK2 (V617F) cells persisted and MPN recurred upon cessation of treatment, suggesting JAK2 inhibitors That May be Unable to Eliminate JAK2 (V617F) cells, Consistent with Preliminary results from Clinical Trials of JAK2 inhibitors in myelofibrosis. …
[3]. Sparidans RW, Durmus S, Xu N, Schinkel AH, Schellens JH, Beijnen JH.Liquid chromatography-tandem mass spectrometric assay for the JAK2 inhibitor CYT387 in plasma.J Chromatogr B Analyt Technol Biomed Life Sci 2012 May 1; 895-896:. 174-7 Epub 2012 Mar 23..
abstract
A quantitative bioanalytical Liquid Chromatography-Tandem Mass spectrometric (LC-MS / MS) assay for the JAK2 inhibitor CYT387 WAS Developed and validated. Plasma samples Were Treated using pre-Protein precipitation with acetonitrile containing cediranib as Internal Standard. The extract WAS Directly Injected into the chromatographic system after dilution with water. This system consisted of a sub-2 μm particle, trifunctional bonded octadecyl silica column with a gradient using 0.005% (v / v) of formic acid in a mixture of water and methanol. The eluate was transferred into the electrospray interface with positive ionization and the analyte was detected in the selected reaction monitoring mode of a triple quadrupole mass spectrometer. The assay was validated in a 0.25-1000 ng / ml calibration range. Within day precisions were 3.0-13.5%, BETWEEN Day Precisions 5.7% and 14.5%. Accuracies Were BETWEEN 96% and 113% for the Whole Calibration range. The Drug WAS stable under All Relevant Analytical Conditions. Finally, the assay successfully WAS Used to ASSESS Drug Levels in mice.
[4] . Monaghan KA, Khong T, Burns CJ, Spencer A.The novel JAK inhibitor CYT387 suppresses Multiple Signalling pathways, and induces apoptosis in Prevents Proliferation phenotypically Diverse myeloma cells.Leukemia 2011 Dec; 25 (12):. 1891-9.
Abstract
Janus kinases (JAKs) are involved in various signalling pathways exploited by malignant cells. In multiple myeloma (MM), the interleukin-6 / JAK / signal transducers and activators of transcription (IL-6 / JAK / STAT) pathway has been the focus of research for a number of years and IL-6 has an established role in MM drug resistance. JAKs therefore make a rational drug target for anti-MM therapy. CYT387 is a novel, orally bioavailable JAK1 / 2 inhibitor, which has recently been described. This preclinical evaluation of CYT387 for treatment of MM demonstrated that CYT387 was able to prevent IL-6-induced phosphorylation of STAT3 and greatly decrease IL-6- and insulin-like growth factor-1-induced phosphorylation of AKT and extracellular signal-regulated kinase in human myeloma cell lines (HMCL). CYT387 inhibited MM proliferation in a time- and dose-dependent manner in 6/8 HMCL, and this was not abrogated by the addition of exogenous IL-6 (3/3 HMCL). Cell cycling was inhibited with a G (2) / M accumulation of cells, and apoptosis was induced by CYT387 in all HMCL tested (3/3). CYT387 synergised in killing HMCL when used in combination with the conventional anti-MM therapies melphalan and bortezomib. Importantly, WAS Also apoptosis induced in Primary Patient MM cells (N = 6) with CYT387 as a single agent, and synergy WAS Seen Again when Combined with Conventional therapies.
[5]. Tyner JW, Bumm TG, Deininger J, Wood L, Aichberger KJ, Loriaux MM, Druker BJ, Burns CJ, Fantino E, Deininger MW.CYT387, a novel JAK2 inhibitor, induces hematologic responses and normalizes inflammatory cytokines in murine myeloproliferative neoplasms.Blood 2010 Jun 24; 115 (25):. 5232- 40. Epub 2010 Apr 12.
Abstract
Activating alleles of Janus kinase 2 (JAK2) SUCH as JAK2 (V617F) are Central to the pathogenesis of myeloproliferative neoplasms (MPN), suggesting Small molecule inhibitors targeting JAK2 That May be therapeutically Useful. We have IDENTIFIED an aminopyrimidine derivative (CYT387), which inhibits JAK1, JAK2, and tyrosine kinase 2 (TYK2) at low nanomolar concentrations, with few additional targets. Between 0.5 and 1.5muM CYT387 caused growth suppression and apoptosis in JAK2-dependent hematopoietic cell lines, while nonhematopoietic cell lines were unaffected. In a murine MPN model, CYT387 normalized white cell counts, hematocrit, spleen size, and restored physiologic levels of inflammatory cytokines. Despite the hematologic responses and reduction of the JAK2 (V617F) allele burden, JAK2 (V617F) cells persisted and MPN recurred upon cessation of treatment, suggesting that JAK2 inhibitors may be unable to eliminate JAK2 (V617F) cells, consistent with preliminary results from clinical trials of JAK2 inhibitors in myelofibrosis. While the clinical benefit of JAK2 inhibitors may be substantial, not the least due to reduction of inflammatory cytokines and symptomatic improvement, our data add to increasing evidence that kinase inhibitor monotherapy of malignant disease is not curative, suggesting a need for drug combinations to optimally target the malignant cells.
Abstract
Somatic mutations in Janus kinase 2 (JAK2), including JAK2V617F, result in dysregulated JAK-signal transducer and activator transcription (STAT) signaling, which is implicated in myeloproliferative neoplasm (MPN) pathogenesis. CYT387 is an ATP-competitive small molecule that potently inhibits JAK1 / JAK2 kinases ( IC (50) = 11 and 18 nM, respectively), with significantly less activity against other kinases, including JAK3 (IC (50) = 155 nM). CYT387 inhibits growth of Ba / F3-JAK2V617F and human erythroleukemia (HEL) cells ( IC (50) approximately 1500 nM) or Ba / F3-MPLW515L cells (IC (50) = 200 nM), but has considerably less activity against BCR-ABL harboring K562 cells (IC = 58 000 nM). Cell lines harboring mutated JAK2 alleles (CHRF-288-11 or Ba / F3-TEL-JAK2) were inhibited more potently than the corresponding pair harboring mutated JAK3 alleles (CMK or Ba / F3-TEL-JAK3), and STAT-5 phosphorylation was inhibited in HEL cells with an IC (50) = 400 nM. …
[2]. Tyner Jeffrey W. et al CYT387, a novel JAK2 inhibitor, induces Hematologic Responses and normalizes inflammatory cytokines in murine myeloproliferative neoplasms Blood June 24, 2010vol. no 115. 255232-5240
Abstract
Activating alleles of Janus kinase 2 (JAK2) SUCH as JAK2 (V617F) are Central to the pathogenesis of myeloproliferative neoplasms (MPN), suggesting Small molecule inhibitors targeting JAK2 That May be therapeutically Useful. IDENTIFIED We have an aminopyrimidine derivative ( CYT387), which inhibits JAK1, JAK2, and tyrosine kinase 2 (TYK2) at low nanomolar concentrations, with few additional targets. Between 0.5 and 1.5muM CYT387 caused growth suppression and apoptosis in JAK2-dependent hematopoietic cell lines, while nonhematopoietic cell lines were unaffected. In a murine MPN model, CYT387 normalized white cell counts, hematocrit, spleen size, and restored physiologic levels of inflammatory cytokines. Despite the hematologic responses and reduction of the JAK2 (V617F) allele burden, JAK2 (V617F) cells persisted and MPN recurred upon cessation of treatment, suggesting JAK2 inhibitors That May be Unable to Eliminate JAK2 (V617F) cells, Consistent with Preliminary results from Clinical Trials of JAK2 inhibitors in myelofibrosis. …
[3]. Sparidans RW, Durmus S, Xu N, Schinkel AH, Schellens JH, Beijnen JH.Liquid chromatography-tandem mass spectrometric assay for the JAK2 inhibitor CYT387 in plasma.J Chromatogr B Analyt Technol Biomed Life Sci 2012 May 1; 895-896:. 174-7 Epub 2012 Mar 23..
abstract
A quantitative bioanalytical Liquid Chromatography-Tandem Mass spectrometric (LC-MS / MS) assay for the JAK2 inhibitor CYT387 WAS Developed and validated. Plasma samples Were Treated using pre-Protein precipitation with acetonitrile containing cediranib as Internal Standard. The extract WAS Directly Injected into the chromatographic system after dilution with water. This system consisted of a sub-2 μm particle, trifunctional bonded octadecyl silica column with a gradient using 0.005% (v / v) of formic acid in a mixture of water and methanol. The eluate was transferred into the electrospray interface with positive ionization and the analyte was detected in the selected reaction monitoring mode of a triple quadrupole mass spectrometer. The assay was validated in a 0.25-1000 ng / ml calibration range. Within day precisions were 3.0-13.5%, BETWEEN Day Precisions 5.7% and 14.5%. Accuracies Were BETWEEN 96% and 113% for the Whole Calibration range. The Drug WAS stable under All Relevant Analytical Conditions. Finally, the assay successfully WAS Used to ASSESS Drug Levels in mice.
[4] . Monaghan KA, Khong T, Burns CJ, Spencer A.The novel JAK inhibitor CYT387 suppresses Multiple Signalling pathways, and induces apoptosis in Prevents Proliferation phenotypically Diverse myeloma cells.Leukemia 2011 Dec; 25 (12):. 1891-9.
Abstract
Janus kinases (JAKs) are involved in various signalling pathways exploited by malignant cells. In multiple myeloma (MM), the interleukin-6 / JAK / signal transducers and activators of transcription (IL-6 / JAK / STAT) pathway has been the focus of research for a number of years and IL-6 has an established role in MM drug resistance. JAKs therefore make a rational drug target for anti-MM therapy. CYT387 is a novel, orally bioavailable JAK1 / 2 inhibitor, which has recently been described. This preclinical evaluation of CYT387 for treatment of MM demonstrated that CYT387 was able to prevent IL-6-induced phosphorylation of STAT3 and greatly decrease IL-6- and insulin-like growth factor-1-induced phosphorylation of AKT and extracellular signal-regulated kinase in human myeloma cell lines (HMCL). CYT387 inhibited MM proliferation in a time- and dose-dependent manner in 6/8 HMCL, and this was not abrogated by the addition of exogenous IL-6 (3/3 HMCL). Cell cycling was inhibited with a G (2) / M accumulation of cells, and apoptosis was induced by CYT387 in all HMCL tested (3/3). CYT387 synergised in killing HMCL when used in combination with the conventional anti-MM therapies melphalan and bortezomib. Importantly, WAS Also apoptosis induced in Primary Patient MM cells (N = 6) with CYT387 as a single agent, and synergy WAS Seen Again when Combined with Conventional therapies.
[5]. Tyner JW, Bumm TG, Deininger J, Wood L, Aichberger KJ, Loriaux MM, Druker BJ, Burns CJ, Fantino E, Deininger MW.CYT387, a novel JAK2 inhibitor, induces hematologic responses and normalizes inflammatory cytokines in murine myeloproliferative neoplasms.Blood 2010 Jun 24; 115 (25):. 5232- 40. Epub 2010 Apr 12.
Abstract
Activating alleles of Janus kinase 2 (JAK2) SUCH as JAK2 (V617F) are Central to the pathogenesis of myeloproliferative neoplasms (MPN), suggesting Small molecule inhibitors targeting JAK2 That May be therapeutically Useful. We have IDENTIFIED an aminopyrimidine derivative (CYT387), which inhibits JAK1, JAK2, and tyrosine kinase 2 (TYK2) at low nanomolar concentrations, with few additional targets. Between 0.5 and 1.5muM CYT387 caused growth suppression and apoptosis in JAK2-dependent hematopoietic cell lines, while nonhematopoietic cell lines were unaffected. In a murine MPN model, CYT387 normalized white cell counts, hematocrit, spleen size, and restored physiologic levels of inflammatory cytokines. Despite the hematologic responses and reduction of the JAK2 (V617F) allele burden, JAK2 (V617F) cells persisted and MPN recurred upon cessation of treatment, suggesting that JAK2 inhibitors may be unable to eliminate JAK2 (V617F) cells, consistent with preliminary results from clinical trials of JAK2 inhibitors in myelofibrosis. While the clinical benefit of JAK2 inhibitors may be substantial, not the least due to reduction of inflammatory cytokines and symptomatic improvement, our data add to increasing evidence that kinase inhibitor monotherapy of malignant disease is not curative, suggesting a need for drug combinations to optimally target the malignant cells.
JAKs are kinases which phosphorylate a group of proteins called Signal Transduction and Activators of Transcription or STATs. When phosphorylated, STATs dimerize, translocate to the nucleus and activate expression of genes which lead to, amongst other things, cellular proliferation.
The central role played by the JAK family of protein tyrosine kinases in the cytokine dependent regulation of both proliferation and end function of several important cell types indicates that agents capable of inhibiting the JAK kinases are useful in the prevention and chemotherapeutic treatment of disease states dependent on these enzymes. Potent and specific inhibitors of each of the currently known four JAK family members will provide a means of inhibiting the action of the cytokines that drive immunological and inflammatory diseases.
Myeloproliferative disorders (MPD) include, among others, polycythemia vera (PV), primary myelofibrosis, thrombocythemia, essential thrombocythemia (ET), idiopathic myelofibrosis (IMF), chronic myelogenous leukemia (CML), systemic mastocystosis (SM), chronic neutrophilic leukemia (CNL), myelodisplastic syndrome (MDS) and systemic mast cell disease (SMCD). JAK2 is a member of the JAK family of kinases in which a specific mutation (JAK2V617F) has been found in 99% of polycythemia vera (PV) patients and 50% of essential thrombocytopenia (ET) and idiopathic myelofibrosis (MF). This mutation is thought to activate JAK2, giving weight to the proposition that a JAK2 inhibitor will be useful in treating these types of diseases.
Asthma is a complex disorder characterized by local and systemic allergic inflammation and reversible airway obstruction. Asthma symptoms, especially shortness of breath, are a consequence to airway obstruction, and death is almost invariably due to asphyxiation. Airway Hyper Responsiveness (AHR), and mucus hyper secretion by goblet cells are two of the principle causes of airway obstruction in asthma patients. Intriguingly recent work in animal experimental models of asthma has underscored the importance of IL-13 as a key player in the pathology of asthma. Using a specific IL-13 blocker, it has been demonstrated that IL-13 acts independently of IL-4 and may be capable of inducing the entire allergic asthma phenotype, without the induction of IgE (i.e. in a non-atopic fashion). This and other models have pointed to an important second tier mechanism for elicitating the pathophysiology of asthma, that is not dependent on the production of IgE by resident B-cells or the presence of eonisophils. A direct induction of AHR by IL-13, represents an important process that is likely to be an excellent target for intervention by new therapies. A contemplated effect of a JAK2 inhibitor to the lungs would result in the suppression of the local release of IL-13 mediated IgE production, and therefore reduction in histaminine release by mast cells and eosinophils. This and other consequences of the absence of IL-13 indicate that many of the effects of asthma may be alleviated through administration of a JAK2 inhibitor to the lungs.
Chronic Obstructive Pulmonary Disease (COPD) is a term which refers to a large group of lung diseases which can interfere with normal breathing. Current clinical guidelines define COPD as a disease state characterized by airflow limitation which is not fully reversible. The airflow limitation is usually both progressive and associated with an abnormal inflammatory response of the lungs to noxious particles and gases, particularly cigarette smoke and pollution. Several studies have pointed to an association between increased production of IL-13 and COPD, lending support to the proposition that the potential alleviation of asthma symptoms by use of a JAK2 inhibitor, may also be achieved in COPD. COPD patients have a variety of symptoms including cough, shortness of breath, and excessive production of sputum. COPD includes several clinical respiratory syndromes including chronic bronchitis and emphysema.
Chronic bronchitis is a long standing inflammation of the bronchi which causes increased production of mucus and other changes. The patient’s symptoms are cough and expectoration of sputum. Chronic bronchitis can lead to more frequent and severe respiratory infections, narrowing and plugging of the bronchi, difficult breathing and disability.
Emphysema is a chronic lung disease which affects the alveoli and/or the ends of the smallest bronchi. The lung loses its elasticity and therefore these areas of the lungs become enlarged. These enlarged areas trap stale air and do not effectively exchange it with fresh air. This results in difficult breathing and may result in insufficient oxygen being delivered to the blood. The predominant symptom in patients with emphysema is shortness of breath.
Additionally, there is evidence of STAT activation in malignant tumors, among them lung, breast, colon, ovarian, prostate and liver cancer, as well as Hodgkins lymphoma, multiple myeloma and hepatocellular carcinoma. Chromosomal translocations involving JAK2 fusions to Tel, Bcr and PCM1 have been described in a number of hematopoietic malignancies including chronic myelogenous leukemia (CML), acute myelogenous leukemia (AML), chronic eosinophilic leukemia (CEL), myelodisplastic syndrome (MDS), myeloproliferative disease (MPD) and acute lymphocytic leukemia (ALL). This suggests treatment of hyperproliferative disorders such as cancers including multiple myeloma; prostate, breast and lung cancer; Hodgkin’s Lymphoma; CML; AML; CEL; MDS; ALL; B-cell Chronic Lymphocytic Leukemia; metastatic melanoma; glioma; and hepatoma, by JAK inhibitors is indicated.
Potent inhibitors of JAK2, in addition to the above, will also be useful in vascular disease such as hypertension, hypertrophy, cardiac ischemia, heart failure (including systolic heart failure and diastolic heart failure), migraine and related cerebrovascular disorders, stroke, Raynaud’s phenomenon, POEMS syndrome, Prinzmetal’s angina, vasculitides, such as Takayasu’s arteritis and Wegener’s granulomatosis, peripheral arterial disease, heart disease and pulmonary arterial hypertension.
Pulmonary arterial hypertension (PAH) is a pulmonary vascular disease affecting the pulmonary arterioles resulting in an elevation in pulmonary artery pressure and pulmonary vascular resistance but with normal or only mildly elevated left-sided filling pressures. PAH is caused by a constellation of diseases that affect the pulmonary vasculature. PAH can be caused by or associated with collagen vascular disorders such as systemic sclerosis (scleroderma), uncorrected congenital heart disease, liver disease, portal hypertension, HIV infection, Hepatitis C, certain toxins, splenectomy, hereditary hemorrhagic teleangiectasia, and primary genetic abnormalities. In particular, a mutation in the bone morphogenetic protein type 2 receptor (a TGF-b receptor) has been identified as a cause of familial primary pulmonary hypertension (PPH). It is estimated that 6% of cases of PPH are familial, and that the rest are “sporadic.” The incidence of PPH is estimated to be approximately 1 case per 1 million population. Secondary causes of PAH have a much higher incidence. The pathologic signature of PAH is the plexiform lesion of the lung which consists of obliterative endothelial cell proliferation and vascular smooth muscle cell hypertrophy in small precapillary pulmonary arterioles. PAH is a progressive disease associated with a high mortality. Patients with PAH may develop right ventricular (RV) failure. The extent of RV failure predicts outcome. The JAK/STAT pathway has recently been implicated in the pathophysiology of PAH. JAKs are kinases which phosphorylate a group of proteins called Signal Transduction and Activators of Transcription or STATs. When phosphorylated, STATs dimerize, translocate to the nucleus and activate expression of genes which lead to proliferation of endothelial cells and smooth muscle cells, and cause hypertrophy of cardiac myocytes. There are three different isoforms of JAK: JAK1, JAK2, and JAK3. Another protein with high homology to JAKs is designated Tyk2. An emerging body of data has shown that the phosphorylation of STAT3, a substrate for JAK2, is increased in animal models of PAH. In the rat monocrotaline model, there was increased phosphorylation of the promitogenic transcription factor STAT3. In this same study pulmonary arterial endothelial cells (PAECs) treated with monocrotaline developed hyperactivation of STAT3. A promitogenic agent or protein is an agent or protein that induces or contributes to the induction of cellular proliferation. Therefore, one effect of JAK2 inhibition would be to decrease proliferation of endothelial cells or other cells, such as smooth muscle cells. A contemplated effect of a JAK2 inhibitor would be to decrease the proliferation of endothelial cells or other cells which obstruct the pulmonary arteriolar lumen. By decreasing the obstructive proliferation of cells, a JAK2 inhibitor could be an effective treatment of PAH.
Additionally the use of JAK kinase inhibitors for the treatment of viral diseases and metabolic diseases is indicated.
Although the other members of the JAK family are expressed by essentially all tissues, JAK3 expression appears to be limited to hematopoetic cells. This is consistent with its essential role in signalling through the receptors for IL-2, IL4, IL-7, IL-9 and IL-15 by non-covalent association of JAK3 with the gamma chain common to these multichain receptors. Males with X-linked severe combined immunodeficiency (XSCID) have defects in the common cytokine receptor gamma chain (gamma c) gene that encodes a shared, essential component of the receptors of interleukin-2 (IL-2), IL-4, IL-7, IL-9, and IL-15. An XSCID syndrome in which patients with either mutated or severely reduced levels of JAK3 protein has been identified, suggesting that immunosuppression should result from blocking signalling through the JAK3 pathway. Gene Knock out studies in mice have suggested that JAK3 not only plays a critical role in B and T lymphocyte maturation, but that JAK3 is constitutively required to maintain T cell function. Taken together with the biochemical evidence for the involvement of JAK3 in signalling events downstream of the IL-2 and IL-4 receptor, these human and mouse mutation studies suggest that modulation of immune activity through the inhibition of JAK3 could prove useful in the treatment of T-cell and B-cell proliferative disorders such as transplant rejection and autoimmune diseases. Conversely undesired inhibition of JAK3 could have a devastating effect on the immune status of an individual treated with drug.
Although the inhibition of various types of protein kinases, targeting a range of disease states, is clearly beneficial, it has been to date demonstrated that the identification of a compound which is selective for a protein kinase of interest, and has good “drug like” properties such as high oral bioavailability, is a challenging goal. In addition, it is well established that the predictability of inhibition, or selectivity, in the development of kinase inhibitors is quite low, regardless of the level sequence similarity between the enzymes being targeted.
The challenges in developing therapeutically appropriate JAK2 inhibitors for use in treatment kinase associated diseases such as immunological and inflammatory diseases including organ transplants; hyperproliferative diseases including cancer and myeloproliferative diseases; viral diseases; metabolic diseases; and vascular diseases include designing a compound with appropriate specificity which also has good drug-likeliness.
There is therefore a continuing need to design and/or identify compounds which specifically inhibit the JAK family of kinases, and particularly compounds which may preferentially inhibit one of the JAK kinases relative to the other JAK kinases, particularly JAK2. There is a need for such compounds for the treatment of a range of diseases.
13
PALBOCICLIB

PALBOCICLIB
Mechanism of action: selective inhibitor of the cyclin-dependent kinases CDK4 and CDK6
Indication: Estrogen receptor-positive (ER+), HER2-negative (HER2 -) breast cancer
Current Status: Phase III (US, UK, EU), (US Clinical trials numbers NCT01864746,NCT01740427, NCT01942135)
Expected Launch Date: 2015
Potential Sales(peak):$5 billion
Company:Pfizer
Indication: Estrogen receptor-positive (ER+), HER2-negative (HER2 -) breast cancer
Current Status: Phase III (US, UK, EU), (US Clinical trials numbers NCT01864746,NCT01740427, NCT01942135)
Expected Launch Date: 2015
Potential Sales(peak):$5 billion
Company:Pfizer
CHEMICAL NAMES
1. Pyrido[2,3-d]pyrimidin-7(8H)-one, 6-acetyl-8-cyclopentyl-5-methyl-2-[[5-(1-
piperazinyl)-2-pyridinyl]amino]-
2. 6-acetyl-8-cyclopentyl-5-methyl-2-{[5-(piperazin-1-yl)pyridin-2-
yl]amino}pyrido[2,3-d]pyrimidin-7(8H)-one
MOLECULAR FORMULA C24H29N7O2
MOLECULAR WEIGHT 447.5
TRADEMARK None as yet
SPONSOR Pfizer Inc.
CODE DESIGNATION PD-0332991
CAS#: 571190-30-2 (PD0332991); 827022-32-2 (PD0332991 HCl salt) 827022-33-3 (palbociclib isethionate)
1. Pyrido[2,3-d]pyrimidin-7(8H)-one, 6-acetyl-8-cyclopentyl-5-methyl-2-[[5-(1-
piperazinyl)-2-pyridinyl]amino]-
2. 6-acetyl-8-cyclopentyl-5-methyl-2-{[5-(piperazin-1-yl)pyridin-2-
yl]amino}pyrido[2,3-d]pyrimidin-7(8H)-one
MOLECULAR FORMULA C24H29N7O2
MOLECULAR WEIGHT 447.5
TRADEMARK None as yet
SPONSOR Pfizer Inc.
CODE DESIGNATION PD-0332991
CAS#: 571190-30-2 (PD0332991); 827022-32-2 (PD0332991 HCl salt) 827022-33-3 (palbociclib isethionate)
http://www.ama-assn.org/resources/doc/usan/palbociclib.pdf FOR STRUCTURE AND DETAILS
recent studies have identified a number of selective CDK4 inhibitors that, as discussed above, may prove useful in treating cancer—either as anti-cancer agents or as chemoprotective agents—and in treating cardiovascular disorders, such as restenosis and atherosclerosis, diseases caused by infectious agents, and autoimmune disorders, including rheumatoid arthritis. For a disclosure of these selective CDK4 inhibitors, see commonly assigned International Patent Application PCT/IB03/00059, filed Jan. 10, 2003 (the ‘059 application), which is herein incorporated by reference in its entirety for all purposes.
The ‘059 application discloses a particularly potent and selective CDK4 inhibitor, 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2,3-d]pyrimidin-7-one:

In standard enzyme assays the compound of Formula 1 exhibits IC50 concentrations for CDK4 and CDK2 inhibition (at 25° C.) of 0.011 μM and >5 μM, respectively. For a discussion of standard CDK4 and CDK2 assays for IC50 determinations, see D. W. Fry et al., J. Biol. Chem. (2001) 16617-16623.
Though the compound of Formula 1 is a potent and selective CDK4 inhibitor, its use in pharmaceutical products presents challenges. For example, the free base has poor water solubility (9 μg/mL) and exhibits low bioavailability in animal studies. A di-HCl salt of the compound of Formula 1 appears to exhibit adequate water solubility. However, moisture uptake studies reveal that, even at low relative humidity (10% RH), the di-HCl salt absorbs water in an amount greater than about 2% of its mass, making it unsuitable for use in a solid drug product. A mono-HCl salt of the compound of Formula 1 is marginally hygroscopic, absorbing more than 2% of its mass at a relative humidity above 80%. However, the process for preparing the mono-HCl salt yields partially crystalline drug substance, indicating potential problems with process scale-up. Other salt forms of the compound of Formula 1 are thus needed.
Pfizer’s breast cancer drug Palbociclib (PD-0332991), a first in the class oral inhibitor of cyclin-dependent kinases (CDK) 4 and 6, is widely seen by investors as Pfizer’s most valuable compound in late-stage development. The FDA awarded Palbociclib “breakthrough therapy designation” in April 2013 based on the preliminary phase 2 data showing palbociclib, combined with Novartis’ drug,Femara (Letrozole), stopped breast tumors progression for more than two years as compared with 7.5 months with letrozole alone. The phase 3 trial started in February 2013 and estimated final completion date is March 2016. Leerink Swann analyst Seamus Fernandez forecasts palbociclib could become a $5 billion drug, with potential for $3 billion in first-line metastatic breast cancer alone.
Palbociclib, also known as PD0332991, is an orally available pyridopyrimidine-derived cyclin-dependent kinase (CDK) inhibitor with potential antineoplastic activity. PD-0332991 selectively inhibits cyclin-dependent kinases (particularly Cdk4/cyclin D1 kinase), which may inhibit retinoblastoma (Rb) protein phosphorylation; inhibition of Rb phosphorylation prevents Rb-positive tumor cells from entering the S phase of the cell cycle (arrest in the G1 phase), resulting in suppression of DNA replication and decreased tumor cell proliferation. PD 0332991 is a highly specific inhibitor of cyclin-dependent kinase 4 (Cdk4) (IC50 = 0.011 μmol/L) and Cdk6 (IC50 = 0.016 μmol/L), having no activity against a panel of 36 additional protein kinases.

6-Acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2,3-d]pyrimidin-7-one hydrochloride (also referred to as “Compound 1”),

as well as its intermediates. Compound 1 is described in U.S. Pat. No. 6,936,612, the disclosure of which is hereby incorporated in its entirety. This compound is a protein kinase inhibitor and represents a synthetic, small molecule inhibitor capable of modulating cell cycle control.
A method of preparing Compound 1 is disclosed as Example 36 of U.S. patent application Ser. No. 6,936,612. Methods of preparing the isethionate salt forms of Compound 1 are disclosed in Examples 1-13 of WO 2005/005426. These methods are for synthesis of small quantities of the salt forms of Compound 1 and are not designed for commercial scale-up. Therefore, a preparation of the salt forms for CDK inhibitor 6-Acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2,3-d]pyrimidin-7-one hydrochloride which is cost-efficient, scaleable and productive is highly desirable.

USAN (zz-153)
PALBOCICLIB ISETHIONATE
THERAPEUTIC CLAIM Antineoplastic
CHEMICAL NAMES
1. Ethanesulfonic acid, 2-hydroxy-, compd. with 6-acetyl-8-cyclopentyl-5-methyl-
2-[[5-(1-piperazinyl)-2-pyridinyl]amino]pyrido[2,3-d]pyrimidin-7(8H)-one (1:1)
THERAPEUTIC CLAIM Antineoplastic
CHEMICAL NAMES
1. Ethanesulfonic acid, 2-hydroxy-, compd. with 6-acetyl-8-cyclopentyl-5-methyl-
2-[[5-(1-piperazinyl)-2-pyridinyl]amino]pyrido[2,3-d]pyrimidin-7(8H)-one (1:1)
2. 6-acetyl-8-cyclopentyl-5-methyl-2-{[5-(piperazin-1-yl)pyridin-2-
yl]amino}pyrido[2,3-d]pyrimidin-7(8H)-one mono(2-hydroxyethanesulfonate)
yl]amino}pyrido[2,3-d]pyrimidin-7(8H)-one mono(2-hydroxyethanesulfonate)
MOLECULAR FORMULA C24H29N7O2 . C2H6O4S
MOLECULAR WEIGHT 573.7
SPONSOR Pfizer, Inc.
CODE DESIGNATIONS PD 0332991-0054, PF-00080665-73
CAS REGISTRY NUMBER 827022-33-3
MOLECULAR WEIGHT 573.7
SPONSOR Pfizer, Inc.
CODE DESIGNATIONS PD 0332991-0054, PF-00080665-73
CAS REGISTRY NUMBER 827022-33-3
- PD 0332991-0054
- PF-00080665-73
- UNII-W1NYL2IRDR
……………………………….




COMPARATIVE EXAMPLE 1A Preparation of 4-[6-(6-bromo-8-cyclopentyl-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino)-pyridin-3-yl]-piperazine-1-carboxylic acid tert-butyl ester
A suspension of 6-bromo-8-cyclopentyl-2-methansulfinyl-5-methyl-8H-pyrido[2,3-d]pyrimidin-7-one (10.00 g, 0.027 mol, prepared as in Example 6 of WO 01/707041, which is incorporated herein by reference) and 10.37 g (0.0373 mol) of 4-(6-amino-pyridin-3-yl)-piperazine-1-carboxylic acid tert-butyl ester in toluene (100 mL) was heated under nitrogen in an oil bath for 7 hours. Thin layer chromatography (SiO2, 10% MeOH/DCM) indicated the presence of both starting materials. The suspension was heated under reflux for an additional 18 hours. The resulting suspension was cooled to RT and filtered to give 4-[6-(6-bromo-8-cyclopentyl-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino)-pyridin-3-yl]-piperazine-1-carboxylic acid tert-butyl ester (5.93 g, 38%). Melting point>250° C. MS (APCI) M++1: calc’d, 584.2, found, 584.2.
COMPARATIVE EXAMPLE 1B Preparation of 4-{6-[8-cyclopentyl-6-(1-ethoxy-vinyl)-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino]-pyridin-3-yl}-piperazine-1-carboxylic acid tert-butyl ester
A suspension of 4-[6-(6-bromo-8-cyclopentyl-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino)-pyridin-3-yl]-piperazine-1-carboxylic acid tert-butyl ester (5.93 g, 0.010 mol, prepared as in Example 1A), tetrakis(triphenylphosphine)palladium(0) (1.40 g, 0.00121 mol), and tributyl(1-ethoxyvinyl)tin (5.32 mL, 0.0157 mol) in toluene (30 mL) was heated under reflux for 3.5 hours. The mixture was cooled and filtered to give a solid. Purification of the solid by silica gel chromatography using a gradient of 5%-66% ethyl acetate/hexane over 15 minutes gave 4-{6-[8-cyclopentyl-6-(1-ethoxy-vinyl)-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino]-pyridin-3-yl}-piperazine-1-carboxylic acid tert-butyl ester as a yellow foam (4.50 g, 78%). MS (APCI) M++1: calc’d 576.2, found, 576.3.
COMPARATIVE EXAMPLE 1C Preparation of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2,3-d]pyrimidin-7-one hydrochloride
Hydrogen chloride gas was bubbled into an ice-bath cooled solution of 4-{6-[8-cyclopentyl-6-(1-ethoxy-vinyl)-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino]-pyridin-3-yl}-piperazine-1-carboxylic acid tert-butyl ester (4.50 g, 0.00783 mol, prepared as in 2005-0059670A1) in DCM (100 mL). The resulting suspension was stoppered and stirred at RT overnight, then diluted with diethyl ether (200 mL). The solid was collected by filtration, washed with diethyl ether, and dried to give the hydrochloride salt of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2,3-d]pyrimidin-7-one as a yellow solid (4.01 g, 92%). Melting point 200° C. HPLC, C18 reverse phase, 10%-95% gradient of 0.1% TFA/CH3CN in 0.1% TFA/H2O during 22 minutes: 99.0% at 11.04 minutes. MS (APCI) M++1: calc’d, 448.2, found, 448.3. Anal. calc’d for C24H29N7O2.2.4H2O.1.85 HCl: C, 51.64; H, 6.44; N, 17.56, Cl (total), 11.75. Found: C, 51.31; H, 6.41; N, 17.20; Cl (total), 12.11.
EXAMPLE 2 Preparation of 4-(6-Nitro-pyridin-3-yl)-piperazine-1-carboxylic acid tert-butyl ester

EXAMPLE 2A Preparation of 4-(6-Nitro-pyridin-3-yl)-piperazine-1-carboxylic acid tert-butyl ester
To 1.0 kg (5 mol) 5-bromo-2-nitropyridine was added 1.2 kg (6.4 mol) boc piperazine (tert-Butyl piperazine-1-carboxylate) in 2.6 L DMSO and 0.5 kg triethylamine under nitrogen. The mixture was heated to 65-70° C. and held for 30 hours after which some solids precipitated. Water was added and the reaction cooled to 25° C. over 2 hrs. The resulting slurry was filtered, washed and dried at 45° C. to give 1.2 kg (79% crude yield) of canary yellow solid intermediate (2A), which was used without further purification in the subsequent step.
EXAMPLE 2 Preparation of 4-(6-Nitro-pyridin-3-yl)-piperazine-1-carboxylic acid tert-butyl ester (2)
60.0 g of 20% Pd(OH)2/C, 1213.1 g (3.9 moles) of intermediate 2a, and isopropanol were charged and stirred in a Parr reactor, then purged under gas, followed by removal of the catalyst under pressure. The filtrates were concentrated in vacuo at ˜20° C. leaving 917 g of dry brown powder (crude yield ˜84%).
EXAMPLE 3 Preparation of 2-Chloro-8-cyclopentyl-5-methyl-8H-pyrido[2,3-d]pyrimidin-7-one

EXAMPLE 3A Preparation of 5-bromo-2-chloro-4-cyclopentyl-aminopyrimidine
To 1 g (0.004 mol) of 5-bromo-2,4-dichloropyrimidine in ethanol was added 1.5 kg (0.018 mol) cyclopentylamine under nitrogen. The mixture was stirred at 25° C. for 2 hrs. Water was added to precipitate the product, and the solid was recrystallized using hexane 4:1 to give a white crystalline product (3A).
EXAMPLE 3 Preparation of 2-Chloro-8-Cyclopentyl-5-methyl-8H-pyrido[2,3-d]pyrimidin-7-one
41.5 g (0.15 mol) of 5-bromo-2-chloro-4-cyclopentylaminopyrimidine 3a and 32.3 g (0.375 mol) of crotonic acid were mixed in 100 L of THF and 105 ml (1.6 mol) diisopropyl ethylamine under nitrogen. The slurry was stirred, evacuated and refilled with nitrogen three times, after which 860 mg (0.0022 mol) palladium dichloride dibenzonitrile complex and 685 mg (0.0022 mol) tri-ortho-tolylphosphine were added and the resulting slurry degassed an additional three times. The mixture was then heated and stirred at 70° C. for 16 hrs, after which 35 ml acetic anhydride was added and the mixture stirred for an additional 1.5 hrs. The mixture was cooled and diluted with 100 ml MTBE and then extracted with 1NHCl, then aqueous sodium bicarbonate and brine. The organic phase was dried over magnesium sulfate, filtered, concentrated in vacuo, and recrystallized from IPA to yield 31.2 g (68%) of crude product (3).
EXAMPLE 4 4-[6-(6-bromo-8-cyclopentyl-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino)-pyridin-3-yl]-piperazine-1-carboxylic acid tert-butyl ester

EXAMPLE 4A Preparation of 2-chloro-8-cyclopentyl-5-methyl-8H-pyrido[2,3-d]pyrimidine-7-one
10 g (0.04 mol) of intermediate 3 and 13 g (0.16 mol) of sodium acetate were mixed with 50 ml of glacial acetic acid and 12 g (0.08 mol) bromine under nitrogen. The solution was heated to 50° C. and stirred for 35 hrs, then cooled to room temperature. Sodium bisulfite solids were added until the bromine color disappeared, then quenched, filtered and washed to provide a solid which was subsequently dissolved in 500 ml hot IPA, filtered hot, and cooled. The resulting crystals were further filtered, and dried in vacuo at 65° C. to yield 8 g (61%) of crude product (4A).
EXAMPLE 4 Preparation of 4-[6-(6-bromo-8-cyclopentyl-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino)-pyridin-3-yl]-piperazine-1-carboxylic acid tert-butyl ester
3.78 g (2.10 equiv; 13.6 mmoles) of intermediate 1, 25 ml toluene and lithium bis(trimethylsilyl)amide in 1 M THF (13.6 mmoles; 13.6 mL; 12.1 g) were mixed for 10 min under nitrogen to form a dark solution. In a separate beaker the intermediate 4a (1.00 equiv, 6.47 mmoles; 2.50 g) was slurried in toluene then added to the mixture containing 1 and stirred for 30 min, after which the combined mixture was quenched with 25 ml 1 M sodium bicarbonate and then filtered. Alternatively, the combined mixture can be quenched with ammonium chloride. The filter cake was washed with toluene, then acetone, then water and dried at 60° C. to give 3.5 g (92%) of a grey-yellow solid 4.
EXAMPLE 5 Preparation of 4-{6-[6-(1-butoxy-vinyl)-8-cycloentyl-5-methyl-7-oxo-7,8-dihydropyrido[2,3-d]pyrimidin-2-ylamino]-pyridin-3-yl}-piperazine-1-carboxylic acid tert-butyl ester

768 g (1.3 mol) of intermediate 4, was mixed with 395 g (3.9 mol) of butyl vinyl ether, 4.7 L of n-butanol, and 275 ml (1.6 mol) diisopropyl ethylamine under nitrogen. The slurry was stirred and placed under ca. 50 tore vacuum and then refilled with nitrogen; this was repeated 2 more times. To this degassed solution was added 22 g (0.03 mol) Bis-(diphenylphosphinoferrocene)palladium dichloride dichloromethane complex and the resulting slurry was degassed an additional three times as described above. The mixture was then heated and stirred at 95° C. for 20 hrs. The resulting thin red slurry was diluted with 4 L branched octane’s and cooled to about 5° C. after which 1 L saturated aq. potassium carbonate was added and the mixture was filtered and rinsed with 500 ml branched octanes. After drying for 16 hrs at 45° C., 664 g (83%) of gray-solid product (5) was obtained. In addition, column chromatography can be used to further purify the crude product.
EXAMPLE 6 Preparation of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2,3-d]pyrimidin-7-one

11.6 g (1.00 eq, 19.2 mmol) of intermediate 5, water (10.1 equiv; 193 mmoles; 3.48 mL; 3.48 g) and methanol (3.62 moles; 146 mL; 116 g) were combined and heated to 55-60° C. Isethionic acid was added slowly until a clear solution was obtained; 3.3 g isethionic acid solution was necessary to reach this end point. The resulting clear orange solution was filtered through paper and rinsed through with 20 ml methanol, after which the filtrate was reheated to 55-60° C. and the remaining isethionic acid was added (a total of 9.93 g was added). The reaction mixture precipitated and thickened for 6 hours, after which it was cooled and held at 30-35° C. while triethylamine (2.92 g; 28.8 mmoles) was added slowly as a 10% solution in methanol over 12 hrs. About halfway through the addition of triethylamine, desired polymorphic seeds were added to help formation of the desired polymorph. The resulting slurry was cooled and held at 5° C. for 15 minutes and the crystals were filtered and washed with methanol. The solid product was dried in vacuo at 55° C. to obtain 11 g of yellow crystals of the title compound.
……………………………………………………………………
EXAMPLES
The following examples are intended to be illustrative and non-limiting, and represent specific embodiments of the present invention.
Example 1 Preparation of 4-[6-(6-bromo-8-cyclopentyl-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino)-pyridin-3-yl]-piperazine-1-carboxylic acid tert-butyl ester
A suspension of 6-bromo-8-cyclopentyl-2-methansulfinyl-5-methyl-8H-pyrido[2,3-d]pyrimidin-7-one (10.00 g, 0.027 mol, prepared as in Example 6 of WO 01/707041, which is incorporated herein by reference) and 10.37 g (0.0373 mol) of 4-(6-amino-pyridin-3-yl)-piperazine-1-carboxylic acid tert-butyl ester in toluene (100 mL) was heated under nitrogen in an oil bath for 7 hours. Thin layer chromatography (SiO2, 10% MeOH/DCM) indicated the presence of both starting materials. The suspension was heated under reflux for an additional 18 hours. The resulting suspension was cooled to RT and filtered to give 4-[6-(6-bromo-8-cyclopentyl-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino)-pyridin-3-yl]-piperazine-1-carboxylic acid tert-butyl ester (5.93 g, 38%). Melting point>250° C. MS (APCI) M++1: calc’d, 584.2, found, 584.2.
Example 2 Preparation of 4-{6-[8-cyclopentyl-6-(1-ethoxy-vinyl)-5-methyl-7-oxo-7,8-dihydro-pyrido[2.3-d]pyrimidin-2-ylamino]-pyridin-3-yl}-piperazine-1-carboxylic acid tert-butyl ester
A suspension of 4-[6-(6-bromo-8-cyclopentyl-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino)-pyridin-3-yl]-piperazine-1-carboxylic acid tert-butyl ester (5.93 g, 0.010 mol, prepared as in Example 1), tetrakis(triphenylphosphine)palladium(0) (1.40 g, 0.00121 mol), and tributyl(1-ethoxyvinyl)tin (5.32 mL, 0.0157 mol) in toluene (30 mL) was heated under reflux for 3.5 hours. The mixture was cooled and filtered to give a solid. Purification of the solid by silica gel chromatography using a gradient of 5%-66% ethyl acetate/hexane over 15 minutes gave 4-{6-[8-cyclopentyl-6-(1-ethoxy-vinyl)-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino]-pyridin-3-yl}-piperazine-1-carboxylic acid tert-butyl ester as a yellow foam (4.50 g, 78%). MS (APCI) M++1: calc’d 576.2, found, 576.3.
Example 3 Preparation of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2,3-d]pyrimidin-7-one hydrochloride
Hydrogen chloride gas was bubbled into an ice-bath cooled solution of 4-{6-[8-cyclopentyl-6-(1-ethoxy-vinyl)-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino]-pyridin-3-yl}-piperazine-1-carboxylic acid tert-butyl ester (4.50 g, 0.00783 mol, prepared as in Example 2) in DCM (100 mL). The resulting suspension was stoppered and stirred at RT overnight, then diluted with diethyl ether (200 mL). The solid was collected by filtration, washed with diethyl ether, and dried to give the hydrochloride salt of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2,3-d]pyrimidin-7-one as a yellow solid (4.01 g, 92%). Melting point 200° C. HPLC, C18 reverse phase, 10%-95% gradient of 0.1% TFA/CH3CN in 0.1% TFA/H2O during 22 minutes: 99.0% at 11.04 minutes. MS (APCI) M++1: calc’d, 448.2, found, 448.3. Anal. calc’d for C24H29N7O2.2.4H2O.1.85 HCl: C, 51.64; H, 6.44; N, 17.56, Cl (total), 11.75. Found: C, 51.31; H, 6.41; N, 17.20; Cl (total), 12.11.
Example 4 Preparation of a mono-isethionate salt of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2.3-d]pyrimidin-7-one (Form B)
To a slurry of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2,3-d]pyrimidin-7-one (7.0 g, 15.64 mmol, prepared as in Example 3 following contact with NaOH) dispersed in 250 mL of water was added drop-wise 30 mL of a 0.52 M solution of isethionic acid in MeOH (15.64 mmol) to a pH of 5.2. The solution was filtered through a glass filter (fine) and the clear solution was freeze-dried to give 9.4 g of the amorphous salt. The amorphous salt (3.16 g) was mixed with 25 mL of MeOH and after almost complete dissolution a new precipitate formed. Another 25 mL of MeOH was added and the mixture was stirred at 46° C. to 49° C. for four hours. The mixture was slowly cooled to 32° C. and put in a cold room (+4° C.) overnight. A sample was taken for PXRD, which indicated formation of Form B. The mixture was filtered and the precipitate was dried overnight at 50° C. in a vacuum oven. This furnished 2.92 g of the mono-isethionate salt of the compound of Formula 1 in 92% yield. HPLC-99.25%, PXRD-Form B, CHNS, H-NMR were consistent with the structure.
Example 5 Preparation of a mono-isethionate salt of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2.3-d]pyrimidin-7-one (Form B)
MeOH (100 mL) was placed in a 250 mL flask equipped with a mechanical stirrer, thermocouple/controller, condenser, and heating mantle and preheated to 35° C. An amorphous isethionate salt (2 g, prepared as in Example 4) was slowly added in three even portions with a 25 min to 30 min interval between the additions. The reaction mixture was stirred overnight at 35° C. and subsequently cooled. A sample was filtered and examined by PXRD. It was pure Form B. The whole reaction mixture was then used as Form B seeds in a larger scale experiment.
Example 6 Preparation of a mono-isethionate salt of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2,3-d]pyrimidin-7-one (Form B)
MeOH (50 mL) was placed in a 250 mL flask equipped with a magnetic stirrer, condenser, thermocouple/controller, and heating mantle, and preheated to 40° C. An amorphous isethionate salt (1 g, prepared as in Example 4) was slowly added in three even portions with 30 min interval between the portions and then stirred overnight at 40° C. The reaction was monitored by in-situ Raman spectroscopy. The sample was taken, filtered and analyzed by PXRD. It was pure Form B by PXRD and Raman spectroscopy. The mixture was cooled to 25° C. at a rate of 3° C./h, cooled to −10° C., filtered, and vacuum dried to furnish 0.85 g of the Form B crystalline product.
Example 7 Preparation of a mono-isethionate salt of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2,3-d]pyrimidin-7-one (Form B)
The free base (Formula 1, 0.895 mg, 2 mmol) was mixed with 10 mL of MeOH and seeded with 33 mg of a mono-isethionate salt of the compound of Formula 1 (Form B). Then 5.6 mL of a 0.375 M solution of isethionic acid in MeOH (2.1 mmol) was added in 10 even portions over 75 min time period. The mixture was stirred for an additional hour and a sample was taken for PXRD analysis. It confirmed formation of crystalline Form B. The mixture was stirred at RT overnight and another PXRD was taken. There was no change in the crystal form. The mixture was cooled in a refrigerator at −8° C. overnight, filtered, and dried at 50° C. in a vacuum oven to give 1.053 g (91.8% of theory) of the above-named compound (Form B). HPLC—99.8%, CHNS, H-NMR, IR are consistent with the structure, PXRD-Form B.
Example 8 Preparation of a mono-isethionate salt of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2.3-d]pyrimidin-7-one (Form A)
An amorphous isethionate salt (47 mg, prepared as in Example 4) was mixed with 4 mL of EtOH in a 15 mL flask equipped with a magnetic stirrer, thermocouple and condenser. The mixture was heated to reflux, which resulted in the formation of a nearly clear solution. After refluxing for 10-15 min, the mixture became cloudy. It was slowly cooled to 50° C. and was seeded at 69° C. with Form A. The mixture was held at 50° C. for 5 h and was allowed to cool to RT overnight. The mixture was subsequently cooled to 1° C. with an ice bath, held for 1.5 h, filtered, washed with 0.5 mL of cold EtOH, air-dried, and then dried in a vacuum oven at 70° C. overnight to furnished 38.2 mg of a fine crystalline material. The crystalline material was found to be mono-isethionate salt Form A by PXRD. H-NMR was consistent for the mono-isethionate salt and indicated the presence of residual EtOH ca. 5.9 mol % or 0.6 wt %.
Example 9 Preparation of a mono-isethionate salt of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-pyrido[2,3-d]pyrimidin-7-one (Form D)
An amorphous isethionate salt (9.0 g, prepared as in Example 4) was mixed with 300 mL of MeOH, stirred and heated to 63.8° C. (at reflux). To the slightly cloudy mixture was added two 50-mL portions of MeOH. The hot mixture was filtered into a 2-L flask equipped with a mechanical stirrer. The mixture was briefly heated to reflux and then cooled to 60° C. IPA (100 mL) was added to the mixture. The mixture was again heated to 60° C. and an additional 110 mL of IPA was added. A precipitate started to form at 59.7° C. The mixture was reheated to 67.5° C., cooled to 50° C., and held overnight. A sample was taken the next morning for PXRD analysis. The mixture was cooled to 25° C. at a rate of 3° C./h and another PXRD sample was taken when the mixture reached 28° C. The mixture was allowed to cool to RT overnight. A precipitate was collected and dried in a vacuum oven at 65° C. and 30 Torr. The procedure produced 7.45 g (82.8% yield) of the crystalline compound (Form D by PXRD analysis). Previously analyzed samples were also Form D. HPLC showed 98.82% purity and CHNS microanalysis was within +/−0.4%. A slurry of isethionate salt Form A, B, and D in MeOH yielded substantially pure Form B in less than three days.
Example 10 Preparation of isethionic acid (2-hydroxy-ethanesulfonic acid)
A 5-L, four-necked, round-bottomed flask, equipped with mechanical stirrer, thermocouple, gas sparger, and an atmosphere vent through a water trap was charged with 748 g (5.05 mol) of sodium isethionate (ALDRICH), and 4 L of IPA. The slurry was stirred at RT. An ice bath was used to keep the internal temperature below 50° C. as 925 g (25.4 mol) of hydrogen chloride gas (ALDRICH) was sparged into the system at a rate such that it dissolved as fast as it was added (as noted by lack of bubbling through the water trap). Sufficient HCl gas was added until the system was saturated (as noted by the start of bubbling through the water trap). During the addition of HCl, the temperature rose to 45° C. The slurry was cooled to RT and filtered over a coarse-fritted filter. The cake was washed with 100 mL of IPA and the cloudy filtrate was filtered through a 10-20μ filter. The resulting clear, colorless filtrate was concentrated under reduced pressure on a rotary evaporator, while keeping the bath temperature below 50° C. The resulting 1.07 kg of clear, light yellow oil was diluted with 50 mL of tap water and 400 mL of toluene and concentrated under reduced pressure on a rotary evaporator for three days, while keeping the bath temperature below 50° C. The resulting 800 g of clear, light yellow oil was diluted with 500 mL of toluene and 250 mL of IPA and concentrated under reduced pressure on a rotary evaporator for 11 days, keeping the bath temperature below 50° C. The resulting 713 g of clear, light yellow oil was titrated at 81 wt % (580 g, 91.1% yield) containing 7.9 wt % water and 7.5 wt % IPA.
Example 11 Preparation of 4-{6-[6-(1-butoxy-vinyl)-8-cyclopentyl-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino]-pyridin-3-yl}-piperazine-1-carboxylic acid tert-butyl ester
A 5-L, three-necked, round-bottomed flask, equipped with a mechanical stirrer, a thermocouple, and a nitrogen inlet/outlet vented through a silicone oil bubbler was placed under a nitrogen atmosphere and charged with 4-[6-(6-bromo-8-cyclopentyl-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino)-pyridin-3-yl]-piperazine-1-carboxylic acid tert-butyl ester (300 g, 0.51 mol, prepared as in Example 2), butyl vinyl ether (154 g, 1.54 mol, ALDRICH), n-butanol (1.5 L, ALDRICH), and diisopropyl ethylamine (107 mL, 0.62 mol, ALDRICH). The slurry was placed under approximately 50 Torr vacuum and then refilled with nitrogen 3 times. To this was added 8.3 g (0.01 mol) bis-(diphenylphosphinoferrocene) palladium dichloride dichloromethane (JOHNSON MATTHEY, Lot 077598001) and the resulting slurry was purged an additional three times as described above. The mixture was then heated to 95° C. and stirred for 20 h. The resulting thin red slurry was diluted with 2 L of heptane and cooled to approximately 5° C. At this temperature, 400 mL saturated aqueous potassium carbonate was added and the mixture was filtered and rinsed with 250 mL of heptane. After drying in an oven for 16 h at 45° C., 231.7 g (75% yield) of the title compound was obtained as a yellow solid.
Example 12 Preparation of a mono-isethionate salt of 6-acetyl-8-cyclopentyl-5-methyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-8H-Pyrido[2,3-d]pyrimidin-7-one (Form B)
A 22-L, three-necked, round-bottomed flask, equipped with a mechanical stirrer, a thermocouple, and a nitrogen inlet/outlet vented through a silicone oil bubbler was placed under a nitrogen atmosphere and charged with 4-{6-[6-(1-butoxy-vinyl)-8-cyclopentyl-5-methyl-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidin-2-ylamino]-pyridin-3-yl}-piperazine-1-carboxylic acid tert-butyl ester (725 g, 1.20 mol, prepared as in Example 11) and MeOH (14 L). The slurry was stirred at RT as it was charged with a solution of isethionic acid (530 g, 4.20 mol, prepared as in Example 10), MeOH (1.5 L), and water (70 mL, 3.89 mol). The resulting slurry was heated to 55° C. over 30 minutes and then stirred at 55° C. for 30 minutes. A solution of 175 g (1.73 mol) of Et3N (ALDRICH) in 200 mL of MeOH was charged to the slurry as it was cooled to 30° C. The slurry was held at 30° C. as a solution of 128 g (1.26 mol) of Et3N in 2 L of MeOH was added dropwise over 6 hours. The resulting slurry was sampled to determine crystal form (Form B). The slurry was cooled and held at 5° C. for 15 minutes and was subsequently filtered through a coarse-fritted filter. The resulting filter cake was washed with multiple washes of 200 mL of cold MeOH. The solid product was dried at 55° C. under vacuum to yield 710 g (91% yield) of the title compound as yellow crystals.
1)Peter L. Toogood, Patricia J. Harvey, Joseph T. Repine, Derek J. Sheehan, Scott N. VanderWel, Hairong Zhou, Paul R. Keller, Dennis J. McNamara, Debra Sherry, Tong Zhu, Joanne Brodfuehrer, Chung Choi, Mark R. Barvian, and David W. Fry;Discovery of a Potent and Selective Inhibitor of Cyclin-Dependent Kinase 4/6; Journal of Medicinal Chemistry, 2005, 48(7),2388-2406;
2)Scott N. VanderWel, Patricia J. Harvey, Dennis J. McNamara, Joseph T. Repine, Paul R. Keller, John Quin III, R. John Booth, William L. Elliott, Ellen M. Dobrusin, David W. Fry, and Peter L. Toogood; Pyrido[2,3-d]pyrimidin-7-ones as Specific Inhibitors of Cyclin-Dependent Kinase 4; Journal of Medicinal Chemistry,2005,48(7),2371-2387;
3)Erdman, David Thomas et al;Preparation of 2-(pyridin-2-ylamino)-pyrido[2,3-d]pyrimidin-7-ones;PCT Int. Appl., WO2008032157
4)Sharpless, Norman E. et al;Hematopoietic protection against chemotherapeutic compounds using selective cyclin-dependent kinase 4/6 inhibitors;PCT Int. Appl., WO2010039997
5)Dirocco, Derek Paul et al;Protection of renal tissues from schema through inhibition of the proliferative kinases CDK4 and CDK6;PCT Int. Appl., WO2012068381
6)Logan, Joshua E.et al.;PD- 0332991, a potent and selective inhibitor of cyclin-dependent kinase 4/6, demonstrates inhibition of proliferation in renal cell carcinoma at nanomolar concentrations and molecular markers predict for sensitivity; Anticancer Research (2013), 33(8), 2997-3004.
7)Phase III Study Evaluating Palbociclib (PD-0332991), a Cyclin-Dependent Kinase (CDK) 4/6 Inhibitor in Patients With Hormone-receptor-positive, HER2-normal Primary Breast Cancer With High Relapse Risk After Neoadjuvant Chemotherapy “PENELOPEB”;ClinicalTrials.gov number:NCT01864746;currently recruiting participants(as of January 2, 2013)
8)A Randomized, Multicenter, Double-Blind Phase 3 Study Of PD-0332991 (Oral CDK 4/6 Inhibitor) Plus Letrozole Versus Placebo Plus Letrozole For The Treatment Of Postmenopausal Women With ER (+), HER2 (-) Breast Cancer Who Have Not Received Any Prior Systemic Anti Cancer Treatment For Advanced Disease;ClinicalTrials.gov number:NCT01740427;currently recruiting participants(as of January 2, 2013)
9)Multicenter, Randomized, Double-Blind, Placebo-Controlled, Phase 3 Trial Of Fulvestrant (Faslodex®) With Or Without PD-0332991 (Palbociclib) +/- Goserelin In Women With Hormone Receptor-Positive, HER2-Negative Metastatic Breast Cancer Whose Disease Progressed After Prior Endocrine Therapy;ClinicalTrials.gov number:NCT01942135;currently recruiting participants(as of January 2, 2013)
| US6936612 | Jan 16, 2003 | Aug 30, 2005 | Warner-Lambert Company | 2-(Pyridin-2-ylamino)-pyrido[2,3-d]pyrimidin-7-ones |
| WO2005005426A1 | Jun 28, 2004 | Jan 20, 2005 | Vladimir Genukh Beylin | Isethionate salt of a selective cdk4 inhibitor |
| US20030229026 * | Dec 18, 2000 | Dec 11, 2003 | Al-Awar Rima Salim | Agents and methods for the treatment of proliferative diseases |
| US20040006074 * | Dec 2, 2002 | Jan 8, 2004 | The Government Of The United States Of America | Cyclin dependent kinase (CDK)4 inhibitors and their use for treating cancer |
| US20040048915 * | Sep 24, 2001 | Mar 11, 2004 | Engler Thomas Albert | Methods and compounds for treating proliferative diseases |
| US20050222163 * | Mar 30, 2005 | Oct 6, 2005 | Pfizer Inc | Combinations of signal transduction inhibitors |
| US20070027147 * | Dec 3, 2004 | Feb 1, 2007 | Takashi Hayama | Biarylurea derivatives |
| WO2008032157A2 * | Aug 27, 2007 | Mar 20, 2008 | David Thomas Erdman | Synthesis of 2-(pyridin-2-ylamino)-pyrido[2,3-d]pyrimidin-7-ones |
| WO2010075074A1 | Dec 15, 2009 | Jul 1, 2010 | Eli Lilly And Company | Protein kinase inhibitors |
| WO2012098387A1 | Jan 17, 2012 | Jul 26, 2012 | Centro Nacional De Investigaciones Oncológicas (Cnio) | 6, 7-ring-fused triazolo [4, 3 - b] pyridazine derivatives as pim inhibitors |
| US7781583 | Sep 10, 2007 | Aug 24, 2010 | Pfizer Inc | Synthesis of 2-(pyridin-2-ylamino)-pyrido[2,3-d] pryimidin-7-ones |
| US7855211 | Dec 15, 2009 | Dec 21, 2010 | Eli Lilly And Company | Protein kinase inhibitors |
| US8247408 * | Oct 9, 2006 | Aug 21, 2012 | Exelixis, Inc. | Pyridopyrimidinone inhibitors of PI3Kα for the treatment of cancer |
| US8273755 | Feb 9, 2010 | Sep 25, 2012 | Pfizer Inc | 4-methylpyridopyrimidinone compounds |
old info
Date: April 10, 2013
Pfizer Inc. said that its experimental pill for advanced, often deadly breast cancer has been designated as a breakthrough therapy by the Food and Drug Administration.
The breakthrough designation, created under legislation enacted last summer to fund and improve operations of the FDA, is meant to speed up development and review of experimental treatments that are seen as big advances over existing therapies for serious diseases. Pfizer is working with the agency to determine exactly what research results it will need to apply for approval of the drug.
Palbociclib is being evaluated as an initial treatment for the biggest subgroup of postmenopausal women whose breast cancer is locally advanced or has spread elsewhere in the body. About 60% of women with such advanced breast cancer have tumors classified as ER+, or estrogen-receptor positive, but HER2-, or lacking an excess of the growth-promoting protein HER2.
Estrogen-receptor positive tumors have proteins inside and on the surface of their cells to which the estrogen hormone can attach and then fuel growth of cells. These tumors tend to grow slowly and can be fought with drugs that block estrogen’s effects.
Meanwhile, about 80% of breast cancer tumor cells are HER2 negative. That means that unlike HER2 positive tumors, they don’t produce too much of the HER2 protein, which makes tumors grow and spread more aggressively than in other breast cancer types.
New York-based Pfizer is currently running a late-stage study of palbociclib at multiple centers, comparing its effects when used in combination with letrozole with the effects of letrozole alone.
Letrozole, sold under the brand name Femara for about the past 15 years, is a pill that works by inhibiting aromatase. That’s an enzyme in the adrenal glands that makes estrogen.
According to Pfizer, palbociclib targets enzymes called cyclin dependent kinases 4 and 6. By inhibiting those enzymes, the drug has been shown in laboratory studies to block cell growth and suppress copying of the DNA of the cancer cells.
Pfizer, which has made research on cancer medicines a priority in recent years, also is testing palbociclib as a treatment for other cancers.
| Highlight of recent study using PD-0332991 |
Phase I study of PD-0332991: Forty-one patients were enrolled. DLTs were observed in five patients (12%) overall; at the 75, 125, and 150 mg once daily dose levels. The MTD and recommended phase II dose of PD 0332991 was 125 mg once daily. Neutropenia was the only dose-limiting effect. After cycle 1, grade 3 neutropenia, anemia, and leukopenia occurred in five (12%), three (7%), and one (2%) patient(s), respectively. The most common non-hematologic adverse events included fatigue, nausea, and diarrhea. Thirty-seven patients were evaluable for tumor response; 10 (27%) had stable disease for ≥4 cycles of whom six derived prolonged benefit (≥10 cycles). PD 0332991 was slowly absorbed (median T(max), 5.5 hours), and slowly eliminated (mean half-life was 25.9 hours) with a large volume of distribution (mean, 2,793 L). The area under the concentration-time curve increased linearly with dose. Using an E(max) model, neutropenia was shown to be proportional to exposure. CONCLUSIONS:
PD 0332991 warrants phase II testing at 125 mg once daily, at which dose neutropenia was the sole significant toxicity. (Source: Clin Cancer Res; 18(2); 568-76.)
PD 0332991 warrants phase II testing at 125 mg once daily, at which dose neutropenia was the sole significant toxicity. (Source: Clin Cancer Res; 18(2); 568-76.)
Phase I study of PD-0332991 in 3-week cycles (Schedule 2/1): Six patients had DLTs (18%; four receiving 200 mg QD; two receiving 225 mg QD); the MTD was 200 mg QD. Treatment-related, non-haematological adverse events occurred in 29 patients (88%) during cycle 1 and 27 patients (82%) thereafter. Adverse events were generally mild-moderate. Of 31 evaluable patients, one with testicular cancer achieved a partial response; nine had stable disease (≥10 cycles in three cases). PD 0332991 was slowly absorbed (mean T(max) 4.2 h) and eliminated (mean half-life 26.7 h). Volume of distribution was large (mean 3241 l) with dose-proportional exposure. Using a maximum effective concentration model, neutropenia was proportional to exposure. CONCLUSION: PD 0332991 was generally well tolerated, with DLTs related mainly to myelosuppression. The MTD, 200 mg QD, is recommended for phase II study. (source: Br J Cancer. 2011 Jun 7;104(12):1862-8)
| References |
1: Flaherty KT, Lorusso PM, Demichele A, Abramson VG, Courtney R, Randolph SS, Shaik MN, Wilner KD, O’Dwyer PJ, Schwartz GK. Phase I, dose-escalation trial of the oral cyclin-dependent kinase 4/6 inhibitor PD 0332991, administered using a 21-day schedule in patients with advanced cancer. Clin Cancer Res. 2012 Jan 15;18(2):568-76. doi: 10.1158/1078-0432.CCR-11-0509. Epub 2011 Nov 16. PubMed PMID: 22090362.
2: Smith D, Tella M, Rahavendran SV, Shen Z. Quantitative analysis of PD 0332991 in mouse plasma using automated micro-sample processing and microbore liquid chromatography coupled with tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2011 Oct 1;879(27):2860-5. doi: 10.1016/j.jchromb.2011.08.009. Epub 2011 Aug 16. PubMed PMID: 21889427.
3: Katsumi Y, Iehara T, Miyachi M, Yagyu S, Tsubai-Shimizu S, Kikuchi K, Tamura S, Kuwahara Y, Tsuchiya K, Kuroda H, Sugimoto T, Houghton PJ, Hosoi H. Sensitivity of malignant rhabdoid tumor cell lines to PD 0332991 is inversely correlated with p16 expression. Biochem Biophys Res Commun. 2011 Sep 16;413(1):62-8. doi: 10.1016/j.bbrc.2011.08.047. Epub 2011 Aug 17. PubMed PMID: 21871868; PubMed Central PMCID: PMC3214763.
4: Schwartz GK, LoRusso PM, Dickson MA, Randolph SS, Shaik MN, Wilner KD, Courtney R, O’Dwyer PJ. Phase I study of PD 0332991, a cyclin-dependent kinase inhibitor, administered in 3-week cycles (Schedule 2/1). Br J Cancer. 2011 Jun 7;104(12):1862-8. doi: 10.1038/bjc.2011.177. Epub 2011 May 24. PubMed PMID: 21610706; PubMed Central PMCID: PMC3111206.
5: Nguyen L, Zhong WZ, Painter CL, Zhang C, Rahavendran SV, Shen Z. Quantitative analysis of PD 0332991 in xenograft mouse tumor tissue by a 96-well supported liquid extraction format and liquid chromatography/mass spectrometry. J Pharm Biomed Anal. 2010 Nov 2;53(3):228-34. doi: 10.1016/j.jpba.2010.02.031. Epub 2010 Feb 26. PubMed PMID: 20236782.
6: Finn RS, Dering J, Conklin D, Kalous O, Cohen DJ, Desai AJ, Ginther C, Atefi M, Chen I, Fowst C, Los G, Slamon DJ. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009;11(5):R77. doi: 10.1186/bcr2419. PubMed PMID: 19874578; PubMed Central PMCID: PMC2790859.
7: Menu E, Garcia J, Huang X, Di Liberto M, Toogood PL, Chen I, Vanderkerken K, Chen-Kiang S. A novel therapeutic combination using PD 0332991 and bortezomib: study in the 5T33MM myeloma model. Cancer Res. 2008 Jul 15;68(14):5519-23. doi: 10.1158/0008-5472.CAN-07-6404. PubMed PMID: 18632601.
8: Fry DW, Harvey PJ, Keller PR, Elliott WL, Meade M, Trachet E, Albassam M, Zheng X, Leopold WR, Pryer NK, Toogood PL. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther. 2004 Nov;3(11):1427-38. PubMed PMID: 15542782.
14
SORAFENIB

Sorafenib
(4-(4-(3-(4-chloro-3-(trifluoromethyl)phenyl)ureido)phenoxy)-N-methylpicolinamide)
BAY 43-9006

Bayer HealthCare has obtained approval from the Japanese Ministry of Health, Labour and Welfare (MHLW) for its Nexavar (sorafenib) for treatment of patients with unresectable differentiated thyroid carcinoma.
Bayer HealthCare has obtained approval from the Japanese Ministry of Health, Labour and Welfare (MHLW) for its Nexavar (sorafenib) for treatment of patients with unresectable differentiated thyroid carcinoma.
Nexavar’s approval in Japan is supported by data from the multicentre, placebo-controlled Phase III DECISION (‘stuDy of sorafEnib in loCally advanced or metastatIc patientS with radioactive Iodine refractory thyrOid caNcer’) study.
The international Phase III DECISION study, which randomised a total of 417 patients, met its primary endpoint of extended progression-free survival. Safety and tolerability profile of sorafenib was generally consistent with the known profile of sorafenib.
The most common treatment-emergent adverse events in the sorafenib arm were hand-foot skin reaction, diarrhea, alopecia, weight loss, fatigue, hypertension and rash.
Nexavar was awarded orphan drug status by the MHLW for thyroid carcinoma in September 2013.
Sorafenib (co-developed and co-marketed by Bayer and Onyx Pharmaceuticals as Nexavar),[1] is a drug approved for the treatment of primary kidney cancer (advanced renal cell carcinoma), advanced primary liver cancer (hepatocellular carcinoma), and radioactive iodine resistant advanced thyroid carcinoma.
Medical uses
At the current time sorafenib is indicated as a treatment for advanced renal cell carcinoma (RCC), unresectable hepatocellular carcinomas (HCC) and thyroid cancer.[2][3][4][5]
Kidney cancer
An article in The New England Journal of Medicine, published January 2007, showed compared with placebo, treatment with sorafenib prolongs progression-free survival in patients with advanced clear cell renal cell carcinoma in whom previous therapy has failed. The median progression-free survival was 5.5 months in the sorafenib group and 2.8 months in the placebo group (hazard ratio for disease progression in the sorafenib group, 0.44; 95% confidence interval [CI], 0.35 to 0.55; P<0.01).[6] A few reports described patients with stage IV renal cell carcinomas that were successfully treated with a multimodal approach including neurosurgical, radiation, and sorafenib.[7] This is one of two TGA-labelled indications for sorafenib, although it is not listed on the Pharmaceutical Benefits Scheme for this indication.[5][8]
Liver cancer
At ASCO 2007, results from the SHARP trial[9] were presented, which showed efficacy of sorafenib in hepatocellular carcinoma. The primary endpoint was median overall survival, which showed a 44% improvement in patients who received sorafenib compared to placebo (hazard ratio 0.69; 95% CI, 0.55 to 0.87; p=0.0001). Both median survival and time to progression showed 3-month improvements. There was no difference in quality of life measures, possibly attributable to toxicity of sorafenib or symptoms related to underlying progression of liver disease. Of note, this trial only included patients with Child-Pugh Class A (i.e. mildest) cirrhosis. The results of the study appear in the July 24, 2008, edition of The New England Journal of Medicine. Because of this trial Sorafenib obtained FDA approval for the treatment of advanced hepatocellular carcinoma in November 2007.[10]
In a randomized, double-blind, phase II trial combining sorafenib with doxorubicin, the median time to progression was not significantly delayed compared with doxorubicin alone in patients with advanced hepatocellular carcinoma. Median durations of overall survival and progression-free survival were significantly longer in patients receiving sorafenib plus doxorubicin than in those receiving doxorubicin alone.[10] A prospective single-centre phase II study which included the patients with unresectable hepatocellular carcinoma (HCC)concluding that the combination of sorafenib and DEB-TACE in patients with unresectable HCC is well tolerated and safe, with most toxicities related to sorafenib.[11] This is the only indication for which sorafenib is listed on the PBS and hence the only Government-subsidised indication for sorafenib in Australia.[8] Along with renal cell carcinoma, hepatocellular carcinoma is one of the TGA-labelled indications for sorafenib.[5]
Thyroid cancer
A phase 3 clinical trial has started recruiting (November 2009) to use sorafenib for non-responsive thyroid cancer.[12] The results were presented at the ASCO 13th Annual Meeting and are the base for FDA approval. The Sorafenib in locally advanced or metastatic patients with radioactive iodine-refractory differentiated thyroid cancer: The Phase 3 DECISION trial showed significant improvement in progression-free survival but not in overall survival. However, as is known, the side effects were very frequent, specially hand and foot skin reaction.[13]
Adverse effects
Adverse effects by frequency
Note: Potentially serious side effects are in bold.
Very common (>10% frequency)
Note: Potentially serious side effects are in bold.
Very common (>10% frequency)
- Lymphopenia
- Hypophosphataemia[Note 1]
- Haemorrhage[Note 2]
- Hypertension[Note 3]
- Diarrhea
- Rash
- Alopecia (hair loss; occurs in roughly 30% of patients receiving sorafenib)
- Hand-foot syndrome
- Pruritus (itchiness)
- Erythema
- Increased amylase
- Increased lipase
- Fatigue
- Pain[Note 4]
- Nausea
- Vomiting[Note 5][14]
Common (1-10% frequency)
- Leucopoenia[Note 6]
- Neutropoenia[Note 7]
- Anaemia[Note 8]
- Thrombocytopenia[Note 9]
- Anorexia (weight loss)
- Hypocalcaemia[Note 10]
- Hypokalaemia[Note 11]
- Depression
- Peripheral sensory neuropathy
- Tinnitus[Note 12]
- Congestive heart failure
- Myocardial infarction[Note 13]
- Myocardial ischaemia[Note 14]
- Hoarseness
- Constipation
- Stomatitis[Note 15]
- Dyspepsia[Note 16]
- Dysphagia[Note 17]
- Dry skin
- Exfoliative dermatitis
- Acne
- Skin desquamation
- Arthralgia[Note 18]
- Myalgia[Note 19]
- Renal failure[Note 20]
- Proteinuria[Note 21]
- Erectile dysfunction
- Asthenia (weakness)
- Fever
- Influenza-like illness
- Transient increase in transaminase
Uncommon (0.1-1% frequency)
- Folliculitis
- Infection
- Hypersensitivity reactions[Note 22]
- Hypothyroidism[Note 23]
- Hyperthyroidism[Note 24]
- Hyponatraemia[Note 25]
- Dehydration
- Reversible posteriorleukoencephalopathy
- Hypertensive crisis
- Rhinorrhoea[Note 26]
- Interstitial lung disease-like events[Note 27]
- Gastro-oesophageal reflux disease(GORD)
- Pancreatitis[Note 28]
- Gastritis[Note 29]
- Gastrointestinal perforations[Note 30]
- Increase in bilirubin leading, potentially, to jaundice[Note 31]
- Cholecystitis[Note 32]
- Cholangitis[Note 33]
- Eczema
- Erythema multiforme[Note 34]
- Keratoacanthoma[Note 35]
- Squamous cell carcinoma
- Gynaecomastia (swelling of the breast tissue in men)
- Transient increase in blood alkaline phosphatase
- INR abnormal
- Prothrombin level abnormal
- bulbous skin reaction[15]
Rare (0.01-0.1% frequency)
Mechanism of action
Sorafenib is a small molecular inhibitor of several tyrosine protein kinases (VEGFR and PDGFR) and Raf kinases (more avidly C-Raf than B-Raf).[16][17] Sorafenib also inhibits some intracellular serine/threonine kinases (e.g. C-Raf, wild-type B-Raf and mutant B-Raf).[10] Sorafenib treatment induces autophagy,[18] which may suppress tumor growth. However, autophagy can also cause drug resistance.[19]
History
Renal cancer
Sorafenib was approved by the U.S. Food and Drug Administration (FDA) in December 2005,[20] and received European Commissionmarketing authorization in July 2006,[21] both for use in the treatment of advanced renal cancer.
Liver cancer
The European Commission granted marketing authorization to the drug for the treatment of patients with hepatocellular carcinoma(HCC), the most common form of liver cancer, in October 2007,[22] and FDA approval for this indication followed in November 2007.[23]
In November 2009, the UK’s National Institute of Clinical Excellence declined to approve the drug for use within the NHS in England, Wales and Northern Ireland, stating that its effectiveness (increasing survival in primary liver cancer by 6 months) did not justify its high price, at up to £3000 per patient per month.[24] In Scotland the drug had already been refused authorization by the Scottish Medicines Consortium for use within NHS Scotland, for the same reason.[24]
In March 2012, the Indian Patent Office granted a domestic company, Natco Pharma, a license to manufacture generic Sorafenib, bringing its price down by 97%. Bayer sells a month’s supply, 120 tablets, of Nexavar for 280000 (US$4,700). Natco Pharma will sell 120 tablets for 8800 (US$150), while still paying a 6% royalty to Bayer.[25][26] Under Indian Patents Act, 2005 and the World Trade Organisation TRIPS Agreement, the government can issue a compulsory license when a drug is not available at an affordable price.[27]
280000 (US$4,700). Natco Pharma will sell 120 tablets for 8800 (US$150), while still paying a 6% royalty to Bayer.[25][26] Under Indian Patents Act, 2005 and the World Trade Organisation TRIPS Agreement, the government can issue a compulsory license when a drug is not available at an affordable price.[27]
Thyroid Cancer
As of November 22, 2013, sorafenib has been approved by the FDA for the treatment of locally recurrent or metastatic, progressive differentiated thyroid carcinoma (DTC) refractory to radioactive iodine treatment.[28]
Research
Lung
In some kinds of lung cancer (with squamous-cell histology) sorafenib administered in addition to paclitaxel and carboplatin may bedetrimental to patients.[29]
Brain (Recurrent Glioblastoma)
There is a phase I/II study at the Mayo Clinic[30] of sorafenib and CCI-779 (temsirolimus) for recurrent glioblastoma.
Desmoid Tumor (Aggressive Fibromatosis)
A study performed in 2011 showed that Sorafenib is active against Aggressive fibromatosis. This study is being used as justification for using Sorafenib as an initial course of treatment in some patients with Aggressive fibromatosis.[31]
Nexavar Controversy
In January 2014, Bayer’s CEO stated that Nexavar was developed for “western patients who [could] afford it”. At the prevailing prices, a kidney cancer patient would pay $96,000 (£58,000) for a year’s course of the Bayer-made drug. However, the cost of the Indian version of the generic drug would be around $2,800 (£1,700).[32]
Notes
- Low blood phosphate levels
- Bleeding; including serious bleeds such as intracranial and intrapulmonary bleeds
- High blood pressure
- Including abdominal pain, headache, tumour pain, etc.
- Considered a low (~10-30%) risk chemotherapeutic agent for causing emesis)
- Low level of white blood cells in the blood
- Low level of neutrophils in the blood
- Low level of red blood cells in the blood
- Low level of plasma cells in the blood
- Low blood calcium
- Low blood potassium
- Hearing ringing in the ears
- Heart attack
- Lack of blood supply for the heart muscle
- Mouth swelling, also dry mouth and glossodynia
- Indigestion
- Not being able to swallow
- Sore joints
- Muscle aches
- Kidney failure
- Excreting protein [usually plasma proteins] in the urine. Not dangerous in itself but it is indicative kidney damage
- Including skin reactions and urticaria (hives)
- Underactive thyroid
- Overactive thyroid
- Low blood sodium
- Runny nose
- Pneumonitis, radiation pneumonitis, acute respiratory distress, etc.
- Swelling of the pancreas
- Swelling of the stomach
- Formation of a hole in the gastrointestinal tract, leading to potentially fatal bleeds
- Yellowing of the skin and eyes due to a failure of the liver to adequately cope with the amount of bilirubin produced by the day-to-day actions of the body
- Swelling of the gallbladder
- Swelling of the bile duct
- A potentially fatal skin reaction
- A fairly benign form of skin cancer
- A potentially fatal abnormality in the electrical activity of the heart
- Swelling of the skin and mucous membranes
- A potentially fatal allergic reaction
- Swelling of the liver
- A potentially fatal skin reaction
- A potentially fatal skin reaction
- The rapid breakdown of muscle tissue leading to the build-up of myoglobin in the blood and resulting in damage to the kidneys
 | |
 | |
| SYSTEMATIC (IUPAC) NAME | |
|---|---|
| 4-[4-[[4-chloro-3-(trifluoromethyl)phenyl]carbamoylamino] phenoxy]-N-methyl-pyridine-2-carboxamide | |
| CLINICAL DATA | |
| TRADE NAMES | Nexavar |
| AHFS/DRUGS.COM | monograph |
| MEDLINEPLUS | a607051 |
| LICENCE DATA | EMA:Link, US FDA:link |
| PREGNANCY CAT. | D (AU) D (US) |
| LEGAL STATUS | Prescription Only (S4) (AU) ℞-only (CA) POM (UK) ℞-only (US) |
| ROUTES | Oral |
| PHARMACOKINETIC DATA | |
| BIOAVAILABILITY | 38–49% |
| PROTEIN BINDING | 99.5% |
| METABOLISM | Hepatic oxidation andglucuronidation (CYP3A4 &UGT1A9-mediated) |
| HALF-LIFE | 25–48 hours |
| EXCRETION | Faeces (77%) and urine (19%) |
| IDENTIFIERS | |
| CAS NUMBER | 284461-73-0 |
| ATC CODE | L01XE05 |
| PUBCHEM | CID 216239 |
| DRUGBANK | DB00398 |
| CHEMSPIDER | 187440 |
| UNII | 9ZOQ3TZI87 |
| KEGG | D08524 |
| CHEBI | CHEBI:50924 |
| CHEMBL | CHEMBL1336 |
| SYNONYMS | Nexavar Sorafenib tosylate |
| PDB LIGAND ID | BAX (PDBe, RCSB PDB) |
| CHEMICAL DATA | |
| FORMULA | C21H16ClF3N4O3 |
| MOL. MASS | 464.825 g/mol |

4-(4-{3-[4-chloro-3-(trifluoromethyl)phenyl]ureido}phenoxy)-Λ/2-methylpyridine-2- carboxamide is commonly known as sorafenib (I). Sorafenib is prepared as its tosylate salt. Sorafenib blocks the enzyme RAF kinase, a critical component of the RAF/MEK/ERK signaling pathway that controls cell division and proliferation; in addition, sorafenib inhibits the VEGFR-2/PDGFR-beta signaling cascade, thereby blocking tumor angiogenesis.
Sorafenib, marketed as Nexavar by Bayer, is a drug approved for the treatment of advanced renal cell carcinoma (primary kidney cancer). It has also received “Fast Track” designation by the FDA for the treatment of advanced hepatocellular carcinoma (primary liver cancer). It is a small molecular inhibitor of Raf kinase, PDGF (platelet-derived growth factor), VEGF receptor 2 & 3 kinases and c Kit the receptor for Stem cell factor.

Sorafenib and pharmaceutically acceptable salts thereof is disclosed in WO0042012. Sorafenib is also disclosed in WO0041698. Both these patents disclose processes for the preparation of sorafenib.
WO0042012 and WO0041698 describe the process as given in scheme I which comprises reacting picolinic acid (II) with thionyl chloride in dimethyl formamide (DMF) to form acid chloride salt (III). This salt is then reacted with methylamine dissolved in tetrahydrofuran (THF) to give carboxamide (IV). This carboxamide when further reacted with 4- aminophenol in anhydrous DMF and potassium tert-butoxide 4-(2-(N-methylcarbamoyl)-4- pyridyloxy)aniline (V) is formed. Subsequent reaction of this aniline with 4-chloro-3- (trifluoromethyl) phenyl isocyanate (Vl) in methylene chloride yields sorafenib (I). The reaction is represented by Scheme I as given below.
Scheme I
Picolini

Sorafenib (I)
WO2006034796 also discloses a process for the preparation of sorafenib and its tosylate salt. The process comprises reacting 2-picolinic acid (II) with thionyl chloride in a solvent inert toward thionyl chloride without using dimethyl formamide to form acid chloride salt (III). This acid salt on further reaction with aqueous solution methylamine or gaseous methylamine gives compound (IV). Compound (IV) is then reacted with 4-aminophenol with addition of a carbonate salt in the presence of a base to yield compound (V).
Compound (V) can also be obtained by reacting compound (IV) with 4-aminophenol in the presence of water with addition of a phase transfer catalyst. Compound (V) when reacted with 4-chloro-3-(trifluoromethyl) phenyl isocyanate (Vl) in a non-chlorinated organic solvent, inert towards isocyanate gives sorafenib (I). Sorafenib by admixing with p- toluenesulfonic acid in a polar solvent gives sorafenib tosylate (VII). The reaction is represented by Scheme Il as given below.
Scheme Il
P

A key step in the synthesis of sorafenib is the formation of the urea bond. The processes disclosed in the prior art involve reactions of an isocyanate with an amine. These isocyanate compounds though commercially available are very expensive. Further synthesis of isocyanate is very difficult which requires careful and skillful handling of reagents.
Isocyanate is prepared by reaction of an amine with phosgene or a phosgene equivalent, such as bis(trichloromethyl) carbonate (triphosgene) or trichloromethyl chloroformate (diphosgene). Isocyanate can also be prepared by using a hazardous reagent such as an azide. Also, the process for preparation of an isocyanate requires harsh reaction conditions such as strong acid, higher temperature etc. Further, this isocyanate is reacted with an amine to give urea.
Reactions of isocyanates suffer from one or more disadvantages. For example phosgene or phosgene equivalents are hazardous and dangerous to use and handle on a large scale. These reagents are also not environment friendly. Isocyanates themselves are thermally unstable compounds and undergo decomposition on storage and they are incompatible with a number of organic compounds. Thus, the use of isocyanate is not well suited for industrial scale application.
Sorafenib and its pharmaceutically acceptable salts and solvates are reported for the first time in WO0041698 (corresponding US 03139605) by Bayer. In the literature only one route is disclosed for the preparation of sorafenib. According to this route (Scheme-I), picolinic acid of formula III is reacted with thionyl chloride to give the 4-chloro derivative which on treatment

VII
Scheme-I with methanol gave the methyl ester of formula V. Compound of formula V is reacted with methylamine to get the corresponding amide of formula VL Compound of formula VI is reacted with 4-aminophenol to get the ether derivative of formula VII. Compound of formula VII is reacted with 4-chloro-3-trifluoromethylphenylisocyante to get sorafenib base of formula I. Overall yield of sorafenib in this process is 10% from commercially available 2-picolinic acid of formula II. Main drawback in this process is chromatographic purification of the intermediates involved in the process and low yield at every step.
Donald Bankston’s (Org. Proc. Res. Dev., 2002, 6, 777-781) development of an improved synthesis of the above basic route afforded sorafenib in an overall yield of 63% without involving any chromatographic purification. Process improvements like reduction of time in thionyl chloride reaction; avoid the isolation of intermediates of formulae IV and V5 reduction of base quantity in p-aminophenol reaction, etc lead to the simplification of process and improvement in yield of final compound of formula I.
Above mentioned improvements could not reduce the number of steps in making sorafenib of formula-I. In the first step all the raw materials are charged and heated to target temperature (72°C). Such a process on commercial scale will lead to sudden evolution of gas emissions such as sulfur dioxide and hydrogen chloride. Also, in the aminophenol reaction two bases (potassium carbonate and potassium t-butoxide) were used in large excess to accomplish the required transformation.
A scalable process for the preparation of sorafenib is disclosed in WO2006034796. In this process also above mentioned chemistry is used in making sorafenib of formula I. In the first step, catalytic quantity. of DMF used in the prior art process is replaced with reagents like hydrogen bromide, thionyl bromide and sodium bromide. Yield of required product remained same without any advantages from newly introduced corrosive reagents. Process improvements like change of solvents, reagents, etc were applied in subsequent steps making the process scalable. Overall yield of sorafenib is increased to 74% from the prior art 63% yield. Purity of sorafenib is only 95% and was obtained as light brown colored solid.
Main drawbacks in this process are production of low quality sorafenib and requirement of corrosive and difficult to handle reagents such as thionyl bromide and hydrogen bromide. Also, there is no major improvement in the yield of sorafenib.
Sorafenib tosylate ( Brand name: Nexavar ®, BAY 43-9006 other name, Chinese name: Nexavar, sorafenib, Leisha Wa) was Approved by U.S. FDA for the treatment of advanced kidney cancer in 2005 and liver cancer in 2007 .
Sorafenib, co-Developed and co-marketed by Germany-based Bayer AG and South San Francisco-based Onyx Pharmaceuticals , is an Oral Multi-kinase inhibitor for VEGFR1, VEGFR2, VEGFR3, PDGFRbeta, Kit, RET and Raf-1.
In March 2012 Indian drugmaker Natco Pharma received the first compulsory license ever from Indian Patent Office to make a generic Version of Bayer’s Nexavar despite the FACT that Nexavar is still on Patent. This Decision was based on the Bayer Drug being too expensive to most patients. The Nexavar price is expected to drop from $ 5,500 per person each month to $ 175, a 97 percent decline. The drug generated $ 934 million in global sales in 2010, according to India’s Patent Office.

Sorafenib tosylate
Chemical Name: 4-Methyl-3-((4 – (3-pyridinyl)-2-pyrimidinyl) amino)-N-(5 – (4-methyl-1H-imidazol-1-yl) -3 – (trifluoromethyl) phenyl) benzamide monomethanesulfonate, Sorafenib tosylate
CAS Number 475207-59-1 (Sorafenib tosylate ) , 284461-73-0 (Sorafenib)
References for the Preparation of Sorafenib References
1) Bernd Riedl, Jacques Dumas, Uday Khire, Timothy B. Lowinger, William J. Scott, Roger A. Smith, Jill E. Wood, Mary-Katherine Monahan, Reina Natero, Joel Renick, Robert N. Sibley; Omega-carboxyaryl Substituted diphenyl Ureas as RAF kinase inhibitors ; U.S. Patent numberUS7235576
2) Rossetto, Pierluigi; Macdonald, Peter, Lindsay; Canavesi, Augusto; Process for preparation of sorafenib and Intermediates thereof , PCT Int. Appl., WO2009111061
3) Lögers, Michael; gehring, Reinhold; Kuhn, Oliver; Matthäus, Mike; Mohrs, Klaus; müller-gliemann, Matthias; Stiehl, jürgen; berwe, Mathias; Lenz, Jana; Heilmann, Werner; Process for the preparation of 4 – {4 – [( {[4-chloro-3-(TRIFLUOROMETHYL) phenyl] amino} carbonyl) amino] phenoxy}-N-methylpyridine-2-carboxamide , PCT Int. Appl., WO2006034796
4) Shikai Xiang, Liu Qingwei, Xieyou Rong, sorafenib preparation methods, invention patent application Publication No. CN102311384 , Application No. CN201010212039
5) Zhao multiply there, Chenlin Jie, Xu Xu, MASS MEDIA Ji Yafei; sorafenib tosylate synthesis ,Chinese Journal of Pharmaceuticals , 2007 (9): 614 -616
2) Rossetto, Pierluigi; Macdonald, Peter, Lindsay; Canavesi, Augusto; Process for preparation of sorafenib and Intermediates thereof , PCT Int. Appl., WO2009111061
3) Lögers, Michael; gehring, Reinhold; Kuhn, Oliver; Matthäus, Mike; Mohrs, Klaus; müller-gliemann, Matthias; Stiehl, jürgen; berwe, Mathias; Lenz, Jana; Heilmann, Werner; Process for the preparation of 4 – {4 – [( {[4-chloro-3-(TRIFLUOROMETHYL) phenyl] amino} carbonyl) amino] phenoxy}-N-methylpyridine-2-carboxamide , PCT Int. Appl., WO2006034796
4) Shikai Xiang, Liu Qingwei, Xieyou Rong, sorafenib preparation methods, invention patent application Publication No. CN102311384 , Application No. CN201010212039
5) Zhao multiply there, Chenlin Jie, Xu Xu, MASS MEDIA Ji Yafei; sorafenib tosylate synthesis ,Chinese Journal of Pharmaceuticals , 2007 (9): 614 -616
Preparation of Sorafenib Tosylate (Nexavar) Nexavar, sorafenib Preparation of methyl sulfonate

Sorafenib (Sorafenib) chemical name 4 – {4 – [({[4 - chloro -3 - (trifluoromethyl) phenyl] amino} carbonyl) amino] phenoxy}-N-methyl-pyridine -2 – formamide by Bayer (Bayer) research and development, in 2005 the U.S. Food and Drug Administration (FDA) approval. Trade name Nexavar (Nexavar). This product is an oral multi-kinase inhibitor, for the treatment of liver cancer and kidney cancer.
Indian Patent Office in March this year for Bayer’s Nexavar in liver and kidney cancer drugs (Nexavar) has released a landmark “compulsory licensing” (compulsory license). Indian Patent Office that due to the high price Nexavar in India, the vast majority of patients can not afford the drug locally, thus requiring local Indian pharmaceutical company Natco cheap Nexavar sales. Nexavar in 2017 before patent expiry, Natco pay only Bayer’s pharmaceutical sales to 6% royalties. The move to make Nexavar patent drug prices, the supply price from $ 5,500 per month dropped to $ 175, the price reduction of 97%. Compulsory licensing in India for other life-saving drugs and patent medicines overpriced open a road, the Indian Patent Office through this decision made it clear that the patent monopoly does not guarantee that the price is too high. Nexavar is a fight against advanced renal cell carcinoma, liver cancer cure. In China, a box of 60 capsules of Nexavar price of more than 25,000 yuan. In accordance with the recommended dose, which barely enough to eat half of patients with advanced cancer. In September this year India a patent court rejected Bayer Group in India cheap drugmaker emergency appeal. Indian government to refuse patent medicine overpriced undo “compulsory licensing rules,” allowing the production of generic drugs Nexavar.
Sorafenat by Natco – Sorafenib – Nexavar – India natco Nexavar

Chemical Synthesis of Sorafenib Tosylate (Nexavar)
Sorafenib tosylate (brand name :Nexavar®, other name BAY 43-9006, was approved by US FDA for the treatment of kidney cancer in 2005 and advanced liver cancer in 2007.

US Patent US7235576, WO2006034796, WO2009111061 and Faming Zhuanli Shenqing(CN102311384) disclosed processes for preparation of sorafenib base and its salt sorafenib tosylate.
References
1)Bernd Riedl, Jacques Dumas, Uday Khire, Timothy B. Lowinger, William J. Scott, Roger A. Smith, Jill E. Wood, Mary-Katherine Monahan, Reina Natero, Joel Renick, Robert N. Sibley; Omega-carboxyaryl substituted diphenyl ureas as raf kinase inhibitors; US patent numberUS7235576
2)Rossetto, pierluigi; Macdonald, peter, lindsay; Canavesi, augusto; Process for preparation of sorafenib and intermediates thereof, PCT Int. Appl., WO2009111061
3)Lögers, michael; gehring, reinhold; kuhn, oliver; matthäus, mike; mohrs, klaus; müller-gliemann, matthias; stiehl, jürgen; berwe, mathias; lenz, jana; heilmann, werner; Process for the preparation of 4-{4-[({[4-chloro-3-(trifluoromethyl)phenyl]amino}carbonyl)amino]phenoxy}-n-methylpyridine-2-carboxamide, PCT Int. Appl., WO2006034796CN102311384, CN201010212039
2)Rossetto, pierluigi; Macdonald, peter, lindsay; Canavesi, augusto; Process for preparation of sorafenib and intermediates thereof, PCT Int. Appl., WO2009111061
3)Lögers, michael; gehring, reinhold; kuhn, oliver; matthäus, mike; mohrs, klaus; müller-gliemann, matthias; stiehl, jürgen; berwe, mathias; lenz, jana; heilmann, werner; Process for the preparation of 4-{4-[({[4-chloro-3-(trifluoromethyl)phenyl]amino}carbonyl)amino]phenoxy}-n-methylpyridine-2-carboxamide, PCT Int. Appl., WO2006034796CN102311384, CN201010212039
Full Experimental Details for the preparation of Sorafenib Tosylate (Nexavar)
Synthesis of 4-(2-(N-methylcarbamoyl)-4-pyridyloxy)aniline.
A solution of 4-aminophenol (9.60 g, 88.0 mmol) in anh. DMF (150 mL) was treated with potassium tert-butoxide (10.29 g, 91.7 mmol), and the reddish-brown mixture was stirred at room temp. for 2 h. The contents were treated with 4-chloro- N -methyl-2-pyridinecarboxamide (15.0 g, 87.9mmol) and K2CO3 (6.50 g, 47.0 mmol) and then heated at 80°C. for 8 h. The mixture was cooled to room temp. and separated between EtOAc (500 mL) and a saturated NaCl solution (500 mL). The aqueous phase was back-extracted with EtOAc (300 mL). The combined organic layers were washed with a saturated NaCl solution (4×1000 mL), dried (Na2SO4) and concentrated under reduced pressure. The resulting solids were dried under reduced pressure at 35°C. for 3 h to afford 4-(2-(N-methylcarbamoyl)-4-pyridyloxy)aniline as a light-brown solid 17.9 g, 84%):. 1H-NMR (DMSO-d6) δ 2.77 (d, J = 4.8 Hz, 3H), 5.17 (br s, 2H), 6.64, 6.86 (AA’BB’ quartet, J = 8.4 Hz, 4H), 7.06 (dd, J = 5.5, 2.5 Hz, 1H), 7.33 (d, J = 2.5 Hz, 1H), 8.44 (d, J = 5.5 Hz; 1H), 8.73 (br d, 1H); HPLC ES-MS m/z 244 ((M+H)+).
Synthesis of 4-{4-[({[4-Chloro-3-(trifluoromethyl)phenyl]amino}carbonyl)amino]phenoxy}-N-methylpyridine-2-carboxamide (sorafenib)
4-(4-Aminophenoxy)-N-methyl-2-pyridinecarboxamide (52.3 kg, 215 mol) is suspended in ethyl acetate (146 kg) and the suspension is heated to approx. 40° C. 4-Chloro-3-trifluoromethylphenyl isocyanate (50 kg, 226 mol), dissolved in ethyl acetate (58 kg), is then added to such a degree that the temperature is kept below 60° C. After cooling to 20° C. within 1 h, the mixture is stirred for a further 30 min and the product is filtered off. After washing with ethyl acetate (30 kg), the product is dried under reduced pressure (50° C., 80 mbar). 93 kg (93% of theory) of the title compound are obtained as colorless to slightly brownish crystals. m.p. 206-208° C. 1H-NMR (DMSO-d6, 500 MHz): δ =2.79 (d, J=4.4 Hz, 3H, NCH3); 7.16 (dd, J=2.5, 5.6 Hz, 1H, 5-H); 7.18 (d, J=8.8 Hz, 2H, 3′-H, 5′-H); 7.38 (d, J=2.4 Hz, 1H, 3-H); 7.60-7.68 (m, 4H, 2′-H, 6′-H, 5″-H, 6″-H); 8.13 (d, J=1.9 Hz, 1H, 2″-H); 8.51 (d, J=5.6 Hz, 1H, 6-H); 8.81 (d, J=4.5 Hz, 1H, NHCH3); 9.05 (br. s, 1H, NHCO); 9.25 (br. s, 1H, NHCO) MS (ESI, CH3CN/H2O): m/e=465 [M+H]+.
Synthesis of Sorafenib Tosylate (Nexavar)
4-(4-{3-[4-chloro-3-(trifluoromethyl)phenyl]ureido}phenoxy)-N2-methylpyridine-2-carboxamide (sorafenib) (50g, 0.1076 mol) is suspended in ethyl acetate (500 g) and water (10g). The mixture is heated to 69°C within 0.5 h, and a filtered solution of p-toluenesulfonic acid monohydrate (3.26 g, 0.017 mol) in a mixture of water (0.65 g) and ethyl acetate (7.2 g) is added. After filtration a filtered solution of p-toluenesulfonic acid monohydrate (22g, 0.11 mol) in a mixture of ethyl acetate (48 g) and water (4.34 g) is added. The mixture is cooled to 23°C within 2 h. The product is filtered off, washed twice with ethyl acetate (92.5 g each time) and dried under reduced pressure. The sorafenib tosylate (65.5 g, 96% of theory) is obtained as colorless to slightly brownish crystals.
…………………..
Example 22: Synthesis of Sorafenib
Phenyl 4-chloro-3-(trifluoromethyl)phenylcarbamate (100 g, 0.3174 mol) and 4-(4- aminophenoxy)-N-methylpicolinamide (77.14 g, 0.3174 mol) were dissolved in N1N- dimethyl formamide (300 ml) to obtain a clear reaction mass. The reaction mass was agitated at 40-450C for 2-3 hours, cooled to room temperature and diluted with ethyl acetate (1000 ml). The organic layer was washed with water (250 ml) followed by 1N HCI (250ml) and finally with brine (250 ml). The organic layer was separated, dried over sodium sulfate and degassed to obtain solid. This solid was stripped with ethyl acetate and finally slurried in ethyl acetate (1000 ml) at room temperature. It was then filtered and vacuum dried to give (118 g) of 4-(4-(3-(4-chloro-3- (trifluoromethyl)phenyl)ureido)phenoxy)-N-methylpicolinamide (sorafenib base).
Example 23: Synthesis of 1-(4-chloro-3-(trifluoromethyl)phenyl)urea (Compound 4)
Sodium cyanate (1.7 g, 0.02mol) was dissolved in water (17ml) at room temperature to obtain a clear solution. This solution was then charged drop wise to the clear solution of 3- trifluoromethyl-4-chloroaniline (5 g, 0.025 mol) in acetic acid (25 ml) at 40°C-45°C within 1- 2 hours. The reaction mass was agitated for whole day and cooled gradually to room temperature. The obtained solid was filtered washed with water and vacuum dried at 500C to afford the desired product (5.8 g) i.e. 1-(4-chloro-3-(trifluoromethyl)phenyl)urea.
Example 24: Synthesis of Sorafenib
1-(4-chloro-3-(trifluoromethyl) phenyl)urea (15 g, 0.0628 mol), 1 ,8- diazabicyclo[5.4.0]undec-7-ene (11.75 ml, 0.078 mol) and 4-(4-aminophenoxy)-N- methylpicolinamide (15.27 g, 0.0628 mol) were mixed with dimethyl sulfoxide (45 ml) and the reaction mass was then heated to 110-1200C for 12-18 hours. The reaction mass was cooled to room temperature and quenched in water (250 ml). The quenched mass was extracted repeatedly with ethyl acetate and the combined ethyl acetate layer was then back washed with water. It was dried over sodium sulfate and evaporated under vacuum to obtain solid. The obtained solid was slurried in acetonitrile (150 ml) at ambient temperature and filtered to give 4-(4-(3-(4-chloro-3-(trifluoromethyl) phenyl) ureido) phenoxy)-N-methylpicolinamide (sorafenib base) (17.5 g).
………………………..

EXAMPLES
Example 1
Preparation of l-(4-chloro-3-(trifluoromethyl)phenyI)-3-(4-hydroxyphenyl)urea Into a 250 ml, four-necked RB flask was charged 1O g of 4-aminophenol and 100 ml of toluene. A solution of 4-chloro-3-(trifluoromethyl)phenyl isocyante (20.4 g) in toluene (50 ml) was added to the reaction mass at 25-300C. The reaction mass was stirred at room temperature for 16 h. The reaction mass was filtered and washed the. solid with 50 ml of toluene. The wet material was dried in the oven at 50-60°C to get 29.8 g of title compound as white solid. M.P. is 218-222°C. IR (KBr): 3306, 1673, 1625, 1590, 1560, 1517, 1482, 1435, 1404, 1328, 1261, 1182, 1160, 1146, 1125, 1095, 1032, 884, 849, 832, 812, 766, 746, 724, 683, 539 and 434 cm“1.
Example 2 Preparation of sorafenib tosylate
Into a 100 ml, three-necked RB flask was charged 2.0 g of l-(4-chloro-3- (trifluoromethyl)-phenyl)-3-(4-hydroxyphenyl)urea and 10 ml of DMF. Potassium tert- butoxide (2.3 g) was added to the reaction mass and stirred for 45 min at RT. 4-Chlro-N- methylpicolinamide (1.14 g) and potassium carbonate (0.42 g) were added to the reaction mass and heated to 80°C. The reaction mass was maintained at 80-85°C for 8 h and cooled to 30°C. The reaction mass was poured into water and extracted with ethyl acetate. Ethyl acetate layer was washed with water, brine and dried over sodium sulphate. Solvent was distilled of under reduced pressure.
The crude compound (4.7 g) was dissolved in 10 ml of IPA and added 1.9 g of p- toluenesulfonic acid. The reaction mass was stirred at RT for 15 h and filtered. The wet solid was washed with 10 ml of IPA and dried at 50-60°C to get 3.4 g of title compound as off-white crystalline solid.
…………………..
A Scaleable Synthesis of BAY 43-9006: A Potent Raf Kinase Inhibitor for the Treatment of Cancer
Bayer Research Center, Pharmaceutical Division, 400 Morgan Lane, West Haven, Connecticut 06516, U.S.A.
Org. Proc. Res. Dev., 2002, 6 (6), pp 777–781
DOI: 10.1021/op020205n

Urea 3 (BAY 43-9006), a potent Raf kinase inhibitor, was prepared in four steps with an overall yield of 63%. Significant process research enabled isolation of each intermediate and target without chromatographic purification, and overall yield increases >50% were observed compared to those from previous methods. This report focuses on improved synthetic strategies for production of scaled quantities of 3 for preclinical, toxicological studies. These improvements may be useful to assemble other urea targets as potential therapeutic agents to combat cancer.
Synthesis of N-[4-Chloro-3-(trifluoromethyl)phenyl]({4-[2-(N-methyl-carbamoyl)(4-pyridyloxy)]phenyl}amino)carboxamide (3, BAY 43-9006).
A suspension of 9 (67.00 g, 275.43 mmol) in methylene chloride ———————-DELETE………………………………The solids were washed with methylene chloride (2 × 50 mL) and dried under vacuum for 4 h at 35 °C to afford 3 (118.19 g, 254.27 mmol, 92%) as an off-white solid.
Mp = 210−212 °C.
1H NMR (DMSO-d6, 300 MHz):
δ 2.77 (d, J = 4.8 Hz, 3H, −NHCH3);
7.16 (m, 3H, aromatic);
7.37 (d, J = 2.5 Hz, 1H, aromatic);
7.62 (m, 4H, aromatic);
8.11 (d, J = 2.5 Hz, 1H, aromatic);
8.49 (d, J = 5.5 Hz, 1H, aromatic);
8.77 (br d, 1H, −NHCH3);
8.99 (s, 1H, −NHCO−); 9.21 (s, 1H, −NHCO−).
Mass spectrum (HPLC/ES): m/e = 465 (M + 1).
Anal. Calcd for C21H16N4ClF3O3: C, 54.26; H, 3.47; N, 12.05. Found: C, 54.11; H, 3.49; N, 12.03.
HPLC (ELS) purity >98%: tR = 3.5 min.
Synthesis of N-[4-Chloro-3-(trifluoromethyl)phenyl]({4-[2-(N-methyl-carbamoyl)(4-pyridyloxy)]phenyl}amino)carboxamide (3, BAY 43-9006): Use of CDI.
A solution of 11 (1.25 g, 6.39 mmol) in methylene chloride———————-DELETED……………………. high vacuum at 35 °C for 2 h to afford 3 (2.55 g, 5.49 mmol, 91%) as a white solid. Proton NMR and mass-spectral data were consistent with structure.
Anal. Calcd for C21H16N4ClF3O3: C, 54.26; H, 3.47; N, 12.05; Cl, 7.63. Found: C, 54.24; H, 3.31; N, 12.30; Cl, 7.84.
Mp (differential scanning calorimetry, 10 °C/min): 205.6 °C;
no polymorphs observed.
References
- “FDA Approves Nexavar for Patients with Inoperable Liver Cancer” (Press release). FDA. November 19, 2007. Retrieved November 10, 2012.
- “Nexavar (sorafenib) dosing, indications, interactions, adverse effects, and more”. Medscape Reference. WebMD. Retrieved 26 December 2013.
- “NEXAVAR (sorafenib) tablet, film coated [Bayer HealthCare Pharmaceuticals Inc.]“. DailyMed. Bayer HealthCare Pharmaceuticals Inc. November 2013. Retrieved 26 December 2013.
- “Nexavar 200mg film-coated tablets – Summary of Product Characteristics (SPC) – (eMC)”. electronic Medicines Compendium. Bayer plc. 27 March 2013. Retrieved 26 December 2013.
- “PRODUCT INFORMATION NEXAVAR® (sorafenib tosylate)” (PDF). TGA eBusiness Services. Bayer Australia Ltd. 12 December 2012. Retrieved 26 December 2013.
- Escudier, B; Eisen, T; Stadler, WM; Szczylik, C; Oudard, S; Siebels, M; Negrier, S; Chevreau, C; Solska, E; Desai, AA; Rolland, F; Demkow, T; Hutson, TE; Gore, M; Freeman, S; Schwartz, B; Shan, M; Simantov, R; Bukowski, RM (January 2007). “Sorafenib in advanced clear-cell renal-cell carcinoma”. New England Journal of Medicine 356 (2): 125–34. doi:10.1056/NEJMoa060655.PMID 17215530.
- Walid, MS; Johnston, KW (October 2009). “Successful treatment of a brain-metastasized renal cell carcinoma”. German Medical Science 7: Doc28. doi:10.3205/000087. PMC 2775194. PMID 19911072.
- “Pharmaceutical Benefits Scheme (PBS) -SORAFENIB”. Pharmaceutical Benefits Scheme. Australian Government Department of Health. Retrieved 27 December 2013.
- Llovet, et al. (2008). “Sorafenib in Advanced Hepatocellular Carcinoma” (PDF). New England Journal of Medicine 359 (4): 378–90.
- Keating GM, Santoro A (2009). “Sorafenib: a review of its use in advanced hepatocellular carcinoma”. Drugs 69 (2): 223–40.doi:10.2165/00003495-200969020-00006. PMID 19228077.
- Pawlik TM, Reyes DK, Cosgrove D, Kamel IR, Bhagat N, Geschwind JF (October 2011). “Phase II trial of sorafenib combined with concurrent transarterial chemoembolization with drug-eluting beads for hepatocellular carcinoma”. J. Clin. Oncol. 29 (30): 3960–7. doi:10.1200/JCO.2011.37.1021. PMID 21911714.
- “Phase 3 Trial of Nexavar in Patients With Non-Responsive Thyroid Cancer”
- [1]
- “Chemotherapy-Induced Nausea and Vomiting Treatment & Management”. Medscape Reference. WebMD. 3 July 2012. Retrieved 26 December 2013.
- Hagopian, Benjamin (August 2010). “Unusually Severe Bullous Skin Reaction to Sorafenib: A Case Report”. Journal of Medical Cases 1 (1): 1–3. doi:10.4021/jmc112e. Retrieved 11 February 2014.
- Smalley KS, Xiao M, Villanueva J, Nguyen TK, Flaherty KT, Letrero R, Van Belle P, Elder DE, Wang Y, Nathanson KL, Herlyn M (January 2009). “CRAF inhibition induces apoptosis in melanoma cells with non-V600E BRAF mutations”. Oncogene 28 (1): 85–94. doi:10.1038/onc.2008.362. PMC 2898184. PMID 18794803.
- Wilhelm SM, Adnane L, Newell P, Villanueva A, Llovet JM, Lynch M (October 2008). “Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling”. Mol. Cancer Ther. 7 (10): 3129–40. doi:10.1158/1535-7163.MCT-08-0013. PMID 18852116.
- Zhang Y (Jan 2014). “Screening of kinase inhibitors targeting BRAF for regulating autophagy based on kinase pathways.”. J Mol Med Rep 9 (1): 83–90. PMID 24213221.
- Gauthier A (Feb 2013). “Role of sorafenib in the treatment of advanced hepatocellular carcinoma: An update..”. Hepatol Res 43(2): 147–154. doi:10.1111/j.1872-034x.2012.01113.x. PMID 23145926.
- FDA Approval letter for use of sorafenib in advanced renal cancer
- European Commission – Enterprise and industry. Nexavar. Retrieved April 24, 2007.
- “Nexavar® (Sorafenib) Approved for Hepatocellular Carcinoma in Europe” (Press release). Bayer HealthCare Pharmaceuticals and Onyx Pharmaceuticals. October 30, 2007. Retrieved November 10, 2012.
- FDA Approval letter for use of sorafenib in inoperable hepatocellular carcinoma
- “Liver drug ‘too expensive‘“. BBC News. November 19, 2009. Retrieved November 10, 2012.
- http://www.ipindia.nic.in/ipoNew/compulsory_License_12032012.pdf
- “Seven days: 9–15 March 2012″. Nature 483 (7389): 250–1. 2012. doi:10.1038/483250a.
- “India Patents (Amendment) Act, 2005″. WIPO. Retrieved 16 January 2013.
- [2]
- “Addition of Sorafenib May Be Detrimental in Some Lung Cancer Patients”
- ClinicalTrials.gov NCT00329719 Sorafenib and Temsirolimus in Treating Patients With Recurrent Glioblastoma
- “Activity of sorafenib against desmoid tumor/deep fibromatosis”
- “‘We didn’t make this medicine for Indians… we made it for western patients who can afford it‘“. Daily Mail Reporter. 24 Jan 2014.
External links
- Nexavar.com – Manufacturer’s website
- Prescribing Information – includes data from the key studies justifying the use of sorafenib for the treatment of kidney cancer (particularly clear cell renal cell carcinoma, which is associated with the von Hippel-Lindau gene)
- Patient Information from FDA
- Sorafenib in Treating Patients With Soft Tissue Sarcomas
- Sorafenib Sunitinib differences – diagram
- ClinicalTrials.gov NCT00217399 – Sorafenib and Anastrozole in Treating Postmenopausal Women With Metastatic Breast Cancer
- Cipla launches Nexavar generic at 1/10 of Bayer’s price
| REFERENCE | ||
|---|---|---|
| 1 | * | D. BANKSTON ET AL.: “A Scalable Synthesis of BAY 43-9006: A Potent Raf Kinase Inhibitor for the Treatment of Cancer” ORGANIC PROCESS RESEARCH & DEVELOPMENT, vol. 6, no. 6, 2002, pages 777-781, XP002523918 cited in the application |
| 2 | * | PAN W ET AL: “Pyrimido-oxazepine as a versatile template for the development of inhibitors of specific kinases” BIOORGANIC & MEDICINAL CHEMISTRY LETTERS, PERGAMON, ELSEVIER SCIENCE, GB, vol. 15, no. 24, 15 December 2005 (2005-12-15), pages 5474-5477, XP025314229 ISSN: 0960-894X [retrieved on 2005-12-15] |
| CITING PATENT | FILING DATE | PUBLICATION DATE | APPLICANT | TITLE |
|---|---|---|---|---|
| WO2011036647A1 | Sep 24, 2010 | Mar 31, 2011 | Ranbaxy Laboratories Limited | Process for the preparation of sorafenib tosylate |
| WO2011036648A1 | Sep 24, 2010 | Mar 31, 2011 | Ranbaxy Laboratories Limited | Polymorphs of sorafenib acid addition salts |
| WO2011058522A1 | Nov 12, 2010 | May 19, 2011 | Ranbaxy Laboratories Limited | Sorafenib ethylsulfonate salt, process for preparation and use |
| WO2011092663A2 | Jan 28, 2011 | Aug 4, 2011 | Ranbaxy Laboratories Limited | 4-(4-{3-[4-chloro-3-(trifluoromethyl)phenyl]ureido}phenoxy)-n2-methylpyridine-2-carboxamide dimethyl sulphoxide solvate |
| WO2011113367A1* | Mar 17, 2011 | Sep 22, 2011 | Suzhou Zelgen Biopharmaceutical Co., Ltd. | Method and process for preparation and production of deuterated ω-diphenylurea |
| US8552197 | Nov 12, 2010 | Oct 8, 2013 | Ranbaxy Laboratories Limited | Sorafenib ethylsulfonate salt, process for preparation and use |
| US8604208 | Sep 24, 2010 | Dec 10, 2013 | Ranbaxy Laboratories Limited | Polymorphs of sorafenib acid addition salts |
| US8609854 | Sep 24, 2010 | Dec 17, 2013 | Ranbaxy Laboratories Limited | Process for the preparation of sorafenib tosylate |
| US8618305 | Jan 28, 2011 | Dec 31, 2013 | Ranbaxy Laboratories Limited | Sorafenib dimethyl sulphoxide solvate |
| US8669369 | Mar 17, 2011 | Mar 11, 2014 | Suzhou Zelgen Biopharmaceutical Co., Ltd. | Method and process for preparation and production of deuterated Ω-diphenylurea |
15
NINTEDANIB

NINTEDANIB
656247-17-5
- UNII-G6HRD2P839
- Vargatef
- BIBF 1120
methyl (3Z)-3-{[(4-{methyl[(4-methylpiperazin-1-yl)acetyl]amino}phenyl)amino](phenyl)methylidene}-2-oxo-2,3-dihydro-1H-indole-6-carboxylate

In battle of IPF drugs, BI's nintedanib impresses
As an eagerly-anticipated debate kicks off at the American Thoracic Society conference in San Diego on drugs for idiopathic pulmonary fibrosis, Boehringer Ingelheim has posted promising late-stage data on its offering, nintedanib.
Read more at: http://www.pharmatimes.com/Article/14-05-18/In_battle_of_IPF_drugs_BI_s_nintedanib_impresses.aspx#ixzz32K0f3NqQ
Follow us: @PharmaTimes on Twitter
Follow us: @PharmaTimes on Twitter
Nintedanib, a triple angiokinase inhibitor, is currently being evaluated against advanced HCC in phase I/II clinical trials. Here, we report the underlying molecular mechanism by which nintedanib (BIBF-1120) induces an anti-HCC effect.
NCI: An orally bioavailable, indolinone-derived, receptor tyrosine kinase (RTK) inhibitor with potential antiangiogenic and antineoplastic activities. Multitargeted tyrosine kinase inhibitor BIBF 1120 selectively binds to and inhibits vascular endothelial growth factor receptor (VEGFR), fibroblast growth factor receptor (FGFR) and platelet-derived growth factor receptor (PDGFR) tyrosine kinases, which may result in the induction of endothelial cell apoptosis; a reduction in tumor vasculature; and the inhibition of tumor cell proliferation and migration. In addition, this agent also inhibits members of the Src family of tyrosine kinases, including Src, Lck, Lyn, and FLT-3 (fms-like tyrosine kinase 3). VEGFR, FGFR and PDGFR RTKs play key roles in tumor angiogenesis
Nintedanib (formerly BIBF 1120; trade name Vargatef) is a small molecule of angiokinase inhibitor class inhibiting vascular endothelial growth factor receptor (VEGFR), fibroblast growth factor receptor (FGFR) and platelet derived growth factor receptor(PDGFR) being developed by Boehringer Ingelheim for use as an anti-vascular anti-cancer agent.
Mechanism of action
Nintedanib is an indolinone-derived drug that inhibits the process of blood vessel formation (angiogenesis) in tumours. Angiogenesis inhibitors stop the formation and reshaping of blood vessels in and around tumours, which reduces the tumour’s blood supply, starving tumour cells of oxygen and nutrients leading to cell death and tumour shrinkage. Unlike conventional anti-cancer chemotherapy which has a direct cell killing effect on cancer cells, angiogenesis inhibitors starve the tumour cells of oxygen and nutrients which results in tumour cell death. One of the advantages of this method of anti-cancer therapy is that it is more specific than conventional chemotherapy agents, therefore results in fewer and less severe side effects than conventional chemotherapy.
The process of new blood vessel formation (angiogenesis) is essential for the growth and spread of cancers. It is mediated by signaling molecules (growth factors) released from cancer cells in response to low oxygen levels. The growth factors cause the cells of the tumour’s blood vessel to divide and reorganize resulting in the sprouting of new vessels in and around the tumour, improving its blood supply.
Angiogenesis is a process that is essential for the growth and spread of all solid tumours, blocking it prevents the tumour from growing and may result in tumour shrinkage as well as a reduction in the spread of the cancer to other parts of the body. Nintedanib exerts its anti-cancer effect by binding to and blocking the activation of cell receptors involved in blood vessel formation and reshaping (i.e. VEGFR 1-3, FGFR 1-3 AND PDGFRα and β). Inhibition of these receptors in the cells that make up blood vessels (endothelial cells, smooth muscle cells and pericytes) by Nintedanib leads to programmed cell death, destruction of tumor blood vessels and a reduction in blood flow to the tumour. Reduced tumour blood flow inhibits tumor cell proliferation and migration hence slowing the growth and spread of the cancer.[1]
Adverse effects
Preclinical studies have shown that nintedanib binds in a highly selective manner to the ATP binding domain of its three target receptors, without binding to similarly shaped ATP domains in other proteins, which reduces the potential for undesirable side effects.[2]
The most common side effects observed with nintedanib were reversible elevation in liver enzymes (10-28% of patients) and gastrointestinal disturbance (up to 50%). Side effects observed with nintedanib were worse with the higher 250 mg dose, for this reason subsequent trials have used the equally clinically effective 200 mg dose.[1][2][3][4][5][6][7][8][9]
Nintedanib inhibits the growth and reshaping of blood vessels which is also an essential process in normal wound healing and tissue repair. Therefore a theoretical side effect of nintedanib is reduced wound healing however, unlike other anti-angiogenic agents, this side effect has not been observed in patients receiving nintedanib.
Studies
Preclinical studies have demonstrated that nintedanib selectively binds to and blocks the VEGF, FGF and PDGF receptors, inhibiting the growth of cells that constitute the walls of blood vessels (endothelial and smooth muscle cells and pericytes) in vitro. Nintedanib reduces the number and density of blood vessels in tumours in vivo, resulting in tumour shrinkage.[1][2] Nintedanib also inhibits the growth of cells that are resistant to existing chemotherapy agents in vitro, which suggests a potential role for the agent in patients with solid tumours that are unresponsive to or relapse following current first line therapy.[10]
Early clinical trials of nintedanib have been carried out in patients with non-small cell lung, colorectal, uterine, endometrial, ovarian and cervical cancer and multiple myeloma.[4][5][7][8][9] These studies reported that the drug is active in patients, safe to administer and is stable in the bloodstream. They identified that the maximum tolerated dose of nintedanib is 20 0 mg when taken once a day.

Clinical studies
In the first human trials, nintedanib halted the growth of tumours in up to 50% of patients with non-small cell lung cancer and 76% of patients with advanced colorectal cancer and other solid tumours.[4][8] A complete response was observed in 1/26 patients with non-small cell lung and 1/7 patients with ovarian cancer treated with nintedanib. A further 2 patients with ovarian cancer had partial responses to nintedanib.[8][9]
Two phase II trials have been carried out assessing the efficacy, dosing and side effects of nintedanib in non-small cell lung and ovarian cancer. These trials found that nintedanib delayed relapse in patients with ovarian cancer by two months[6] and that overall survival of patients with non-small cell lung who received nintedanib was similar to that observed with the FDA approved VEGFR inhibitor sorafenib. These trials also concluded that increasing the dose of the nintedanib has no effect on survival.[3]
Current clinical trials
Nintedanib is currently undergoing investigation in phase II and III clinical trials and is yet to be licensed by the FDA. Angiogenesis inhibitors such as nintedanib may be effective in a range of solid tumour types including; lung, ovarian, metastatic bowel, liver and brain cancer.
Several further phase I and II clinical trials with nintedanib are underway. Patients are also being recruited for three phase III clinical trials that will evaluate the potential benefit of nintedanib when added to existing 1st line treatments in patients with ovarian.[11] and 2nd line treatment in non-small cell lung cancer [12][13] The phase III trials of nintedanib in lung cancer have been named LUME-Lung 1 and LUME-Lung 2.
Current phase II trials are investigating the effect of nintedanib in patients with metastatic bowel cancer, liver cancer and the brain tumour: glioblastoma multiforme.[14]
Phase III trials are investigating the use of nintedanib in combination with the existing chemotherapy agents permexetred and docetaxel in patients with non-small cell lung cancer,[15] and in combination with carboplatin and paclitaxel as a first line treatment for patients with ovarian cancer.[16]
A phase III clinical trial is also underway examining the safety and efficacy of nintedanib on patients with the non-cancerous lung condition idiopathic pulmonary fibrosis.[17]
............................
J. Med. Chem., 2009, 52 (14), pp 4466–4480
DOI: 10.1021/jm900431g

Inhibition of tumor angiogenesis through blockade of the vascular endothelial growth factor (VEGF) signaling pathway is a new treatment modality in oncology. Preclinical findings suggest that blockade of additional pro-angiogenic kinases, such as fibroblast and platelet-derived growth factor receptors (FGFR and PDGFR), may improve the efficacy of pharmacological cancer treatment. Indolinones substituted in position 6 were identified as selective inhibitors of VEGF-, PDGF-, and FGF-receptor kinases. In particular, 6-methoxycarbonyl-substituted indolinones showed a highly favorable selectivity profile. Optimization identified potent inhibitors of VEGF-related endothelial cell proliferation with additional efficacy on pericyctes and smooth muscle cells. In contrast, no direct inhibition of tumor cell proliferation was observed. Compounds 2 (BIBF 1000) and 3 (BIBF 1120) are orally available and display encouraging efficacy in in vivo tumor models while being well tolerated. The triple angiokinase inhibitor 3 is currently in phase III clinical trials for the treatment of nonsmall cell lung cancer.
References
- Hilberg, F.; G. J. Roth, M. Krssak, S. Kautschitsch, W. Sommergruber, U. Tontsch-Grunt, P. Garin-Chesa, G. Bader, A. Zoephel, J. Quant, A. Heckel, W. J. Rettig (2008). "BIBF 1120: triple angiokinase inhibitor with sustained receptor blockade and good antitumor efficacy". Cancer Res 68 (12): 4774–82. doi:10.1158/0008-5472.CAN-07-6307. ISSN 1538-7445. PMID 18559524.
- Hilberg, F.; U. Tontsch-Grunt, F. Colbatzky, A. Heckel, R. Lotz, J.C.A. van Meel, G.J. Roth (2004). "BIBF1120 a novel, small molecule triple angiokinase inhibitor: profiling as a clinical candidate for cancer therapy". European Journal of Cancer Supplements 2(50).
- Reck, M.; R. Kaiser, C. Eschbach, M. Stefanic, J. Love, U. Gatzemeier, P. Stopfer, J. von Pawel (2011). "A phase II double-blind study to investigate efficacy and safety of two doses of the triple angiokinase inhibitor BIBF 1120 in patients with relapsed advanced non-small-cell lung cancer". Ann Oncol. ISSN 1569-8041.
- Okamoto, I.; H. Kaneda, T. Satoh, W. Okamoto, M. Miyazaki, R. Morinaga, S. Ueda, M. Terashima, A. Tsuya, A. Sarashina, K. Konishi, T. Arao, K. Nishio, R. Kaiser, K. Nakagawa (2010). "Phase I safety, pharmacokinetic, and biomarker study of BIBF 1120, an oral triple tyrosine kinase inhibitor in patients with advanced solid tumors". Mol Cancer Ther 9 (10): 2825–33. doi:10.1158/1535-7163.MCT-10-0379. ISSN 1538-8514. PMID 20688946.
- Mross, K.; M. Stefanic, D. Gmehling, A. Frost, F. Baas, C. Unger, R. Strecker, J. Henning, B. Gaschler-Markefski, P. Stopfer, L. de Rossi, R. Kaiser (2010). "Phase I study of the angiogenesis inhibitor BIBF 1120 in patients with advanced solid tumors".Clin Cancer Res 16 (1): 311–9. doi:10.1158/1078-0432.CCR-09-0694. ISSN 1078-0432. PMID 20028771.
- Ledermann, J.A. (2009). "A randomised phase II placebo-controlled trial using maintenance therapy to evaluate the vascular targeting agent BIBF 1120 following treatment of relapsed ovarian cancer (OC)". J Clin Oncol 27 (15s): (suppl; abstr 5501).
- Kropff, M.; J. Kienast, G. Bisping, W. E. Berdel, B. Gaschler-Markefski, P. Stopfer, M. Stefanic, G. Munzert (2009). "An open-label dose-escalation study of BIBF 1120 in patients with relapsed or refractory multiple myeloma". Anticancer Res 29 (10): 4233–8.ISSN 1791-7530. PMID 19846979.
- Ellis, P. M.; R. Kaiser, Y. Zhao, P. Stopfer, S. Gyorffy, N. Hanna (2010). "Phase I open-label study of continuous treatment with BIBF 1120, a triple angiokinase inhibitor, and pemetrexed in pretreated non-small cell lung cancer patients". Clin Cancer Res 16 (10): 2881–9. doi:10.1158/1078-0432.CCR-09-2944. ISSN 1078-0432.PMID 20460487.
- du Bois, A.; J. Huober, P. Stopfer, J. Pfisterer, P. Wimberger, S. Loibl, V. L. Reichardt, P. Harter (2010). "A phase I open-label dose-escalation study of oral BIBF 1120 combined with standard paclitaxel and carboplatin in patients with advanced gynecological malignancies". Ann Oncol 21 (2): 370–5. doi:10.1093/annonc/mdp506.ISSN 1569-8041. PMID 19889612.
- Xiang, Q. F.; F. Wang, X. D. Su, Y. J. Liang, L. S. Zheng, Y. J. Mi, W. Q. Chen, L. W. Fu (2011). "Effect of BIBF 1120 on reversal of ABCB1-mediated multidrug resistance".Cell Oncol (Dordr) 34 (1): 33–44. doi:10.1007/s13402-010-0003-7. ISSN 2211-3436.
- "Boehringer Ingelheim - AGO-OVAR 12 / LUME-Ovar 1 Trial Information". 2011.
- "Boehringer Ingelheim - LUME-Lung 2 Trial Information". 2011.
- "Boehringer Ingelheim - LUME-Lung 1 Trial Information". 2011.
- http://clinicaltrials.gov/ct2/results?term=++%09+BIBF+1120&phase=1
- http://clinicaltrials.gov/ct2/show/NCT00805194 Phase III LUME-Lung 1: BIBF 1120 Plus Docetaxel as Compared to Placebo Plus Docetaxel in 2nd Line Non Small Cell Lung Cancer
- http://clinicaltrials.gov/ct2/show/NCT01015118 Phase III BIBF 1120 or Placebo in Combination With Paclitaxel and Carboplatin in First Line Treatment of Ovarian Cancer
- http://clinicaltrials.gov/ct2/show/NCT01335477 Safety and Efficacy of BIBF 1120 at High Dose in Idiopathic Pulmonary Fibrosis Patients II

Ethanesulfonic acid - methyl (3Z)-3-{[(4-{methyl[(4-methyl-1-piperazinyl)acetyl]amino}phenyl)amino](phenyl)methylene}-2-oxo-6-indolinecarboxylate (1:1)
Nintedanib esylate
656247-18-6 [RN]
Methyl (3Z)-3-[({4-[N-methyl-2-(4-methylpiperazin-1-yl)acetamido]phenyl}amino)(phenyl)methylidene]-2-oxo-2,3-dihydro-1H-indole-6-carboxylate ethanesulfonate
Nintedanib esylate [USAN]
(3Z)-2,3-Dihydro-3-[[[4-[methyl[2-(4-methyl-1-piperazinyl)acetyl]amino]phenyl]amino]phenylmethylene]-2-oxo-1H-indole-6-carboxylic acid methyl ester ethanesulfonate
Nintedanib esylate, 656247-18-6, UNII-42F62RTZ4G, , NSC753000, NSC-753000, KB-62821
Molecular Formula: C33H39N5O7S Molecular Weight: 649.75706
16

Lapatinib in 3d

LAPATINIB
CAS : 231277-92-2
CAS Name: N-[3-Chloro-4-[(3-fluorophenyl)methoxy]phenyl]-6-[5[[[2-(methylsulfonyl)ethyl]amino]methyl]-2-furanyl]-4-quinazolinamine
- N-(3-Chloro-4-((3-fluorobenzyl)oxy)phenyl)-6-(5-(((2-(methylsulfonyl)ethyl)amino)methyl)furan-2-yl)quinazolin-4-amine
Manufacturers’ Codes: GW-572016, Smithkline Beecham Corporation
Trademarks: Tykerb (GSK)
Molecular Formula: C29H26ClFN4O4S
Molecular Weight: 581.06
Percent Composition: C 59.94%, H 4.51%, Cl 6.10%, F 3.27%, N 9.64%, O 11.01%, S 5.52%
LAUNCHED 2007

 lapatinib
lapatinib| SYSTEMATIC (IUPAC) NAME | |
|---|---|
| N-[3-chloro-4-[(3-fluorophenyl)methoxy]phenyl]-6- [5-[(2-methylsulfonylethylamino)methyl]-2-furyl] quinazolin-4-amine | |
| CLINICAL DATA | |
| TRADE NAMES | Tykerb, Tyverb |
| AHFS/DRUGS.COM | monograph |
| MEDLINEPLUS | a607055 |
| LICENCE DATA | EMA:Link, US FDA:link |
| PREGNANCY CAT. | |
| LEGAL STATUS | |
| ROUTES | Oral |
| PHARMACOKINETIC DATA | |
| BIOAVAILABILITY | Variable, increased with food |
| PROTEIN BINDING | >99% |
| METABOLISM | Hepatic, mostly CYP3A-mediated (minor 2C19 and2C8 involvement) |
| HALF-LIFE | 24 hours |
| EXCRETION | Mostly fecal |
| IDENTIFIERS | |
| CAS NUMBER | 231277-92-2 388082-78-8 (ditosylate) |
| ATC CODE | L01XE07 |
| PUBCHEM | CID 208908 |
| DRUGBANK | DB01259 |
| CHEMSPIDER | 181006 |
| UNII | 0VUA21238F |
| CHEMICAL DATA | |
| FORMULA | C29H26ClFN4O4S |
| MOL. MASS | 581.058 g/mol |
Lapatinib (INN), used in the form of lapatinib ditosylate, (USAN) (Tykerb/Tyverb, GSK) is an orally active drug for breast cancerand other solid tumours.[1] It is a dual tyrosine kinase inhibitor which interrupts the HER2/neu and epidermal growth factor receptor(EGFR) pathways.[2] It is used in combination therapy for HER2-positive breast cancer. It is used for the treatment of patients with advanced or metastatic breast cancer whose tumors overexpress HER2 (ErbB2).[3]
Status
On March 13, 2007, the U.S. Food and Drug Administration (FDA) approved lapatinib in combination therapy for breast cancer patients already using capecitabine (Xeloda, Roche).[2][3] In January 2010, Tykerb received accelerated approval for the treatment of postmenopausal women with hormone receptor positive metastatic breast cancer that overexpresses the HER2 receptor and for whom hormonal therapy is indicated.[3]
Pharmaceutical company GlaxoSmithKline (GSK) markets the drug under the propriety names Tykerb (mostly US) and Tyverb (mostly Europe).[4] The drug currently has approval for sale and clinical use in the US,[2][4] Australia,[2] Bahrain,[2] Kuwait,[2] Venezuela,[2]Brazil,[5] New Zealand,[5][6] South Korea,[5] Switzerland,[4] Japan, Jordan, the European Union, Lebanon, India and Pakistan.[4]
On the 2nd of August 2013, India's Intellectual Property Appellate Board revoked the patent for Glaxo's Tykerb citing its derivative status, while upholding at the same time the original patent granted for Lapatinib.[7]
The drug lapatinib ditosylate is classified as S/NM (a synthetic compound showing competitive inhibition of the natural product) that is naturally derived or inspired substrate (Gordon M. Cragg, Paul G. Grothaus, and David J. Newman, Impact of Natural Products on Developing New Anti-Cancer Agents, Chem. Rev. 2009, 109, 3012–3043)
Lapatinib ditosylate, an ErB-1 and ErB-2 dual kinase inhibitor, was launched in the U.S. in 2007 for the treatment of advanced or metastatic HER2 (ErbB2) positive breast cancer in women who have received prior therapy, including Herceptin(R) (trastuzumab), in combination with Xeloda(R) (capecitabine). The compound was approved in 2007 in Switzerland and Australia and in 2009 in Canada, for this indication. Regulatory approval has also been obtained in Japan. In December 2007, a positive opinion was received in the E.U. In 2008, the CHMP issued a revised positive opinion confirming the positive benefit-risk profile for lapatinib following review by the CHMP of new data received in February 2008 from GlaxoSmithKline arising from a standard pharmacovigilance evaluation of clinical trial and post-marketing data. The CHMP confirmed that these data do not essentially change the positive benefit-risk profile for lapatinib in its proposed indication. In 2008, the MAA was approved in the E.U. and the product was subsequently commercialized in Germany. In 2009, regulatory applications were filed in the U.S. and the E.U. seeking approval for use of lapatinib as first-line treatment of patients with hormone-sensitive, metastatic (or advanced) breast cancer in combination with anti-hormonal therapy. In 2010, lapatinib was launched on the U.S. market as first-line treatment in combination with Femara(R) to treat hormone positive and HER2-positive advanced breast cancer in postmenopausal women for whom hormonal therapy is indicated. In 2010, the compound was approved and launched in the E.U. for the oral treatment of post-menopausal women with hormone receptor-positive, HER2 (ErbB2) over-expressing metastatic breast cancer and for whom chemotherapy is currently not intended, in combination with an aromatase inhibitor. In 2012, GlaxoSmithKline filed regulatory applications in the U.S. and the E.U. for the oral treatment of patients with HER2 (ErbB2)-positive metastatic breast cancer that has progressed on prior trastuzumab regimens, in combination with trastuzumab. In July 2012, GlaxoSmithKline withdrew this application in the U.S. In 2013, the product was approved for this indication in the E.U.
In terms of clinical development, the National Cancer Institute (US) is currently conducting phase II/III trials for the treatment of bladder cancer. Phase III trials are under way to evaluate the use of lapatinib as first-line treatment of breast cancer. The compound is also being evaluated for several oncologic indications in the treatment of brain, gallbladder, prostate, ovary, endometrium, bladder cancer, cervical and hepatobiliary cancers in collaboration with the National Cancer Institute (NCI). Lapatinib in combination with everolimus is also in early clinical studies for the treatment of lymphoma and non-Hodgkin's lymphoma (NHL). A phase II combination trial is evaluating lapatinib for the treatment of advanced or metastatic colorectal cancer. The National Cancer Institute (NCI) is developing the compound in phase II trials for the treatment of peritoneal cancer, ovarian and ductal carcinoma in situ of the breast (DCIS), while Brown University is conducting combination trials with gemcitabine for the treatment of pancreas metastatic cancer, and Cedars-Sinai Medical Center is conducting phase II clinical trials for treatment for pituitary cancer. Phase III clinical study for the treatment of head and neck was terminated because the study didn´t meet primary endpoint.
Lapatinib was granted fast-track status by the FDA in 2005 for the treatment of refractory advanced or metastatic breast cancer patients who have documented ErbB-2 overexpression and who have failed previous therapy. In 2009, Orphan Drug Designation was received in the U.S. by GlaxoSmithKline for the treatment of ErbB2 positive gastric cancer and for the treatment of ErbB2 positive esophageal cancer.
Breast cancer
Lapatinib is used as a treatment for women's breast cancer in treatment naive, ER+/EGFR+/HER2+ breast cancer patients(now often called "triple positive") and in patients who have HER2-positive advanced breast cancer that has progressed after previous treatment with other chemotherapeutic agents, such as anthracycline, taxane-derived drugs, or trastuzumab (Herceptin, Genentech).
A 2006 GSK-supported randomized clinical trial on female breast cancer previously being treated with those agents (anthracycline, a taxane and trastuzumab) demonstrated that administrating lapatinib in combination with capecitabine delayed the time of further cancer growth compared to regimens that use capecitabine alone. The study also reported that risk of disease progression was reduced by 51%, and that the combination therapy was not associated with increases in toxic side effects.[11] The outcome of this study resulted in a somewhat complex and rather specific initial indication for lapatinib—use only in combination with capecitabine for HER2-positive breast cancer in women whose cancer have progressed following previous chemotherapy with anthracycline, taxanes and trastuzumab.

………………………………………………………..
Patent
Product patent
US6727256
or
........................................................
W09935146 (GSK company, filed on 8 February 1999, I) propose a 2_ chlorine _4_ nitrophenol as the starting material, by addition, catalytic hydrogenation, replace, Suzuki coupling Union, such as reductive amination reaction was prepared by lapatinib

First, the method of the protected aldehyde group, deprotection after the completion of the coupling reaction for the reductive amination reaction, the reaction step so long; due to the use of expensive and highly toxic organic heteroaryl stannane reagent 5 - (_ 1,3-dioxolan-2 - yl) -2 - (tributylstannyl group) _ furan, intermediates for drugs and have greater safety and environmental risks; Furthermore, the process requires the synthesis of intermediate purified by column chromatography, post-processing is more complex.
........................................................
CN102295638A (Qilu Pharmaceutical Co., Ltd., June 24, 2010 application) proposed a method of preparing lapatinib is mixture of 5 - formyl-furan-2 - boronic acid, N-[3 - chloro-4 - [(3 - fluorophenyl) methoxy] phenyl] -6 - iodo-4 - quinazolinamine 2 - methylsulfonyl - ethylamine and the catalyst to the solvent, Mr. into transitional intermediate, and then reducing agent such as sodium triacetoxy borohydride reduction to give the desired product, the synthesis route is as follows:

..........................................................
W02005120504A2 (Glaxo, in June 2005 I filed) proposed an alternative approach: a 4 - chloro-6 - iodine quinazoline as the starting material, with 5 - formyl-furan-2 - boric acid instead of highly toxic tin compounds alkylfuryl prepared lapatinib. The synthetic route is as follows:

............................................................................
Patent
Lapatinib has the structural formula (I) and chemical name N-[3- chloro-4-[(3-fluorophenyl)methoxy]phenyl]-6-[5-[(2-methylsulfonylethylamino)methyl]-2- furyl] quinazolin-4-amine.

BACKGROUND ART
Lapatinib is a tyrosine kinase inhibitor that is used as an orally administered drug as its ditosylate salt to treat certain types of advanced or metastatic breast cancer and other solid tumors. Lapatinib ditosylate was approved by the FDA in 2007 and the EMEA in 2008 and is marketed by GlaxoSmithKline (GSK) under the trade name of Tykerb® in the USA and Tyverb® in Europe.
Lapatinib substance is claimed in US 6,713,485 B2 and US 6,727,256 Bl and lapatinib ditosylate and its crystalline forms are claimed in US 7,157,466 B2. A synthesis of lapatinib that utilises a palladium mediated coupling of a substituted 4-anilino-6-iodo-quinazoline (II) with a 2- (tributylstannyl)furan (Ilia) is disclosed in US 6,727,256 Bl and is also presented in US 7,157,466 B2. In US 7,157,466 B2 a second generation approach was disclosed that utilises a palladium catalysed coupling of a substituted 4-anilino-6-iodo-quinazoline (II) with furan-2-yl-boronic acids (Illb). Following the palladium catalysed coupling reactions utilised in the two synthetic methods of US 6,727,256 Bl and US 7,157,466 B2, only one (US 7,157,466 B2) or two (US 6,727,256 Bl and US 7,157,466 B2) synthetic modification of the structure are utilised before the lapatinib substance is provided (Scheme 1). The EMEA's COMMITTEE FOR MEDICINAL PRODUCTS FOR HUMAN USE (CHMP) has published guidelines titled GUIDELINE ON THE SPECIFICATION LIMITS FOR RESIDUES OF METAL CATALYSTS OR METAL REAGENTS and recommendations are presented for oral exposure to metals, including palladium. For a drug being consumed in quantities not exceeding a 10 g daily dose, a limit of 10 ppm (parts per million) concentration of palladium in the drug substance is recommended. Given this, there is still an unmet need for an alternative synthetic method that can be used for preparation of lapatinib in which the palladium mediated coupling step is performed early in the synthetic route, thereby being capable to provide .

Scheme 1
SUMMARY OF THE INVENTION
There are a number of ways that the levels of a metal, such as palladium, can be controlled in a drug substance through purging of the metal by treatment of the drug substance or its synthetic intermediates or both, including crystallisation, aqueous extraction, filtration through metal absorbent filter aids (Organic Process Research & Development 2005, 9, 198-205), precipitation of the metal from solution, chromatography, and treatment with metal scavenging reagents (Organic Process Research & Development 2003, 7, 733-742). By placing the palladium mediated coupling step downstream in the synthetic route, however, to take advantage of synthetic convergence, the opportunity to reduce the level of palladium in the drug substance is reduced. In contrast, however, by redesigning the synthetic route to move the palladium mediated coupling step upstream, further away from the drug substance, increases the opportunity to control the palladium level in the drug substance. Furthermore, by careful operational design (such as in a precipitation and crystallisation step), the palladium level in the intermediates can be consistently controlled. Given that there is a need, the present invention has addressed these two latter points and utilised them in a novel and efficient process for the manufacture of lapatinib and lapatinib ditosylate.

Scheme 2 - Synthesis of lapatinib and lapatinib ditosylate
In contrast to the prior art methods disclosure in US 6,727,256 Bl and US 7,157,466 B2, the present invention has performed a transition metal catalysed coupling reaction at the most upstream point in the synthetic route based on the utilization of commercially available starting materials SMla (6-iodoquinazolin-4(3H)-one) and SM2a (5-formylfuran-2-ylboronic acid), or their analogues SMI and SM2, to provide IM1. Thus, in one aspect of the present invention, lapatinib is made from a novel compound (IM1) (Scheme 2).
In another aspect of the present invention, a lapatinib ditosylate monohydrate is prepared by crystallizing lapatinib ditosylate in a mixture of water, DMSO and MeCN.
In another aspect of the present invention, novel compound IM1 is synthesized by the cross- coupling of commercially available SMla and SM2a, or their analogues SMI and SM2, in suitable solvents comprised of an organic solvent and water in the presence of a base and a catalyst formed from a transition metal and a ligand (scheme 3).

X = CI, Br, I, OTf Y = CHO, or CH(OR)2
BZ = B(OH)2, B(OR)2, [BF3]M or BR2
Scheme 3
Example
Example 1: Synthesis of 5-(4-oxo-3,4-dihydroquinazolin-6-yl)furan-2-carbaldehyde (IMl)
IM1
A 5:2 v/v mixture of DMSO and H20 (1400 mL) was degassed for 30 min at ambient temperature using nitrogen. 5-Formylfuran-2-ylboronic acid (SM2a; 26.8 g, 193 mmol) was added dissolved in this mixture. [HP(i-Bu)3] BF4 " (840 mg, 2.94 mmol) and Pd(OAc)2 (680 mg, 2.94 mmol) was added and the mixture was stirred at ambient temperature under an atmosphere of nitrogen for 20 min. AcOK (18.8 g, 192 mmol) was added into the reactor and was stirred for 20 min at ambient temperature. 6-Iodoquinazolin-4(3 /)-one (SMla; 40 g, 147 mmol) was added and heated to 80±5°C (internal temperature) in an oil bath under nitrogen, Upon completion of the reaction (HPLC), the reaction mixture was hot-filtered, then hot water (400 mL, 80±5°C) was added into the filtrate. This was slowly cooled to 0-15°C (solid started to precipitate at 70°C (internal temperature) and was then filtered. The filter cake was washed with H20 (80 mL), then with MeCN (60 mL), and dried in vacuo at 60+5°C for 6 h to provide 5-(4-oxo-3,4-dihydroquinazolin-6-yl)-furan-2- carbaldehyde (IMl; 34.6 g, 144 mmol) with 99.7 % HPLC purity in 97.6% HPLC yield. XH NMR (300 MHz, de-DMSO): δ 7.47 (d, / = 3.8 Hz, 1H), 7.69 (d, / = 3.8 Hz, 1H), 7.77 (d, / = 8.6 Hz, 1H), 8.17 (s, 1H), 8.27 (dd, / = 8.6, 2.1 Hz, 1H), 8.52 (d, = 2.1 Hz, 1H), 9.66 (s, 1H); 13C NMR (75 MHz, CDC13): δ 110.5, 122, 6, 123.9, 126.0, 127.5, 129.0, 131.4, 147.1, 150.1, 152.7, 157.6, 161.2, 178,8; ESI-MS, Pos: [M+H]+ mJz 241; IR (cm 1): 1713, 1671, 1604,1462; m.p.: 267°C. See Figure 2 for the DSC/TGA of IMl; See Figure 3 for the X-ray powder diffraction pattern of IMl; Residual concentration of palladium: 230 ppm.
Example 2: Synthesis of 5-(4-chloroquinazolin-6-yl)furan-2-carbaldehyde hydrochloride
(IM2a.HCl)

I 1 reflux IM2a.HCI
Over a 1.5 hour period under an atmosphere of N2, SOCb (86.2 g) in MeCN (145 mL) was added dropwise into a mixture, that had been preheated at reflux for 0.5 h, of IM1 (29 g, 0.121 mol), MeCN (435 mL) and DMF (0.88 g) at reflux. The reaction was terminated when less than 2% (HPLC) of IM1 was remaining. If the reaction did not achieve complete reaction, extra SOCI2was added. The mixture was cooled to about 25±5°C (internal temperature), and was then filtered and washed with MeCN (58 mL) to give ca. 55 g of IM2a.HCl (moist with MeCN) with 82A purity by HPLC. IM2a.HCl: ¾ NMR (300 MHz, d6-DMSO): δ 9.68 (s, 1 H), 9.17 (s, 1H), 8.57 (d, / = 2.0 Hz, 1H), 8.46 (dd, J = 8.6, 2.1 Hz, 1H), 8.02 (d, / = 8.6 Hz, 1H), 7.74 (d, = 3.8 Hz, 1H), 7.60 (d, J = 3.8 Hz, 1H). See Figure 5 for the XH NMR spectrum of IM2a.HCl; 13C NMR (75 MHz, d6- DMSO) δ 179.0, 159. 6, 156.4, 152.9, 149.5, 141.0, 132.6, 129.2, 125.9, 123.2, 122.9, 122.7, 111.5;
IM2a.HCl was purified by column chromatography (eluent: ) to give pure IM2a. IM2a: lH NMR (300 MHz, d6-DMSO): δ 7.53 (d, / = 3.3 Hz, 1H), 7.68 (d, J = 3.3 Hz, 1H), 8.02 (d, / = 8.7 Hz, 1H), 8.42 (d, / = 8.4 Hz, 1H), 8.54 (d, / = 2.1 Hz, 1H), 8.90 (s, 1H), 9.64 (s, 1H); 13C NMR (75 MHz, CDCI3): δ 111.5, 122.8, 122.9, 123.7, 125.9, 129.1, 132.5, 142.1 , 149.3, 152.9, 156.6, 159.7, 179.1.
Example 3: Synthesis of 5-(4-(3-chloro-4-(3-fluorobenzyloxy)phenylamino)
- uinazolin-6-yl)furan-2-carbaldehyde hydrochloride (IM3.HC1)

A mixture of IM2a.HCl (moist with MeCN solvent, prepared from 29 g IM1, 0.120 mol) and 3-chloro-4-(3-fiuorobenzyloxy)aniline (SM3; 27.3 g, 0.108 mol) in MeCN (580 mL) was stirred under reflux, until HPLC analysis showed that the reaction was completed (about 2 h). The mixture was cooled to room temperature (25±5°C), filtered, and washed with MeCN (58 mL). A mixture of the moist crude solid IM3 and THF (870 mL) was treated with a 2.0 N aqueous NaOH (348 mL) and stirred for 3-4 h until most of the solid had dissolved. The mixture was filtered through diatomite and was washed with a saturated aqueous solution of NaCl (87 mL). The organic layer was treated with 10% aqueous HCI (174 mL) and stirred for 0.5 h. The resulting solid was filtered, washed with THF (87 mL), and dried in vacuo at 60+5°C for 16 h to give the crude IM3.HC1 (34 g, 0.067 mol, HPLC purity: 99%).
IM3.HC1: :H NMR (300 MHz, d6-DMSO): δ 9.69 (s, 1H), 9.52 (s, 1H), 8.94 (s, 1H), 8.50 (dd, / = 8.8, 1.7 Hz, 1H), 8.01 (d, / = 8.8 Hz, 1 H), 7.97 (d, J =2.5 Hz, 1H), 7.77 (d, / = 3.8 Hz, 1H), 7.73 (dd, = 9.0, 2.5 Hz, 1H), 7.69 (d, / = 3.8 Hz, 1H), 7.49 (td, 7 = 8.0, 6.1 Hz, 1 H), 7.41-7.28 (m, 3H), 7.20 (td, / = 8.4, 2.2 Hz, 1H), 5.31 (s, 2H).
Free base IM3 is obtained by column chromatography (eluting with EtOAc/DCM, 1:4, v/v). IM3 XH NMR (300 MHz, d6-DMSO): δ 5.28 (s, 2H), 7.19 (td, /= 8.7 Hz, 7 = 2.1 Hz 1H), 7.34 (m, 4H), 7.43 (d, 7 = 3.6 Hz , 1H), 7.49 (m, 1H), 7.73 (dd, 7 = 8.7 Hz 7 = 2.7 Hz, 1H), 7.76 (d, 7 = 3.6 Hz, 1H), 7.88 (d, 7 = 9 Hz, 1H), 8.07 (d, 7 = 2.1 Hz, 1H), 8.32 (dd, 7 = 4.43 Hz, 7 = 1.95 Hz, 1H), 8.95 (d, 7 = 1.5 Hz, 1H), 9.68 (s, 1H).
Example 4: Synthesis of N-(3-chloro-4-(3-fluorobenzyloxy)phenyl)-6-(5-((2- (methylsulfonyl)ethylamino)methyl)furan-2-yl)quinazolin-4-amine ditosylate (lapatinib ditosylate)

I
To a suspension of 2-(methylsulfonyl)ethanamine hydrochloride (SM4.HC1; 12.2 g, 76.7 mmol) in THF (600 mL) was added acetic acid (14.1 g, 235 mmol) followed by DIPEA (30.3 g, 235 mmol) were added. After stirred at ambient temperature for 0.5 h, ¾0 (4.2 g, 233 mmol) and IM3.HC1 (30.0 g, HPLC assay >99%, 58.7 mmol) were added. After being stirred at ambient temperature (20°C) for 4 h, sodium triacetoxyborohydride (37.4 g, 176 mmol) was added and the mixture was stirred at ambient temperature (20°C±5°C; external temperature) until HPLC showed the completion of the reaction. A 10% aqueous solution of sodium hydroxide (90 mL) was added and the mixture was stirred for 30 min. The organic phase was washed with 25% aqueous NH4C1 (60 mL), filtered, treated with -TsOH (40.4 g, 135 mmol) and heated to reflux for 2 h. The mixture was cooled to ambient temperature and stirred for 3 h at ambient temperature. The mixture was filtered, and the filter cake was washed twice with THF (120 mL each) and was then dried under vacuum at 70±5°C for 6 h to give 43 g (46.5 mmol) lapatinib ditosylate with 99.4% HPLC purity.
Lapatinib ditosylate [H NMR (300 MHz, d6-DMSO): δ 11.41(s, 2H), 9.33 (s, 3H), 9.04 (d, / = 1.3 Hz, 2H), 8.93 (s, 2H), 8.41 (dd, J =8.8, 1.6 Hz, 2H), 7.91 (d, J = 2.6 Hz, 2H), 7.54-7.41 (m, 9H), 7.37 - 7.27 (m, 6H), 7.25 (d, / = 3.4 Hz, 2H), 7.22 - 7.13 (m, 2H), 7.08 (dd, / = 8.4, 0.6 Hz, 8H), 6.87 ( d, / = 3.5 Hz, 2H), 5.29 (s, 4H), 4.46 (s, 4H), 3.65 - 3.51 (m, 4H), 3.51 - 3.38 (m, 4H), 2.26 (s, 12H).
A solution of lapatinib ditosylate was converted to its free base form, lapatinib, by washing a solution with aqueous NaOH followed by concentration. Lapatinib: XH NMR (300 MHz, d6-DMSO): δ 2.98 (t, / = 6.75 Hz, 1H), 3.04 (s, 1H), 3.29 (t, J = 6.6 Hz, 1H), 3.83 (s, 1H), 5.28 (s, 1H), 6.50 (d, / = 3.0 Hz, 1H), 7.08 (d, / = 3.3 Hz, 1H), 7.20 (m, 1H), 7.33 (m, 4H), 7.48 (m, 1H), 7.76 (m, 1H), 7.80 (d, 7 = 9 Hz, 1H), 8.04 (d, 7 = 2.75 Hz, 1H), 8.17 (dd, / = 8.7 Hz, / = 1.8 Hz, 1H), 8.56 (s, 1H), 8.75 (d, J = 1.8 Hz, 1H).
Example 5a: Purification of lapatinib ditosylate
Lapatinib ditosylate (5.0 g, 5.4 mmol, 96.5% HPLC purity with the maximum individual impurity at 0.8%) was dissolved in DMSO (10 mL) at 70°C (internal temperature). MeCN (10 mL) was added dropwise into the mixture at 70-80°C (internal temperature) and was stirred at this temperature for 1 h. Over a 4 h period the mixture was cooled to room temperature. MeCN (30 mL) was added dropwise, and the mixture was stirred for lh, then filtered and washed with MeCN (10 mL). The filter cake was dried under vacuum at 60°C for 16 h to give 4.0 g lapatinib ditosylate as crystalline Form 1 (as disclosed in US 7,157,466 B2) with 99.6% HPLC purity in 78% HPLC yield.
Example 5b. Purification of lapatinib ditosylate.
Lapatinib ditosylate (3 g, 3.25 mmol, 99.3% HPLC purity was dissolved in DMF (18 mL) at 80°C and stirred for 1 hour. The mixture was hot-filtered. MeCN (18 mL) was added into the filtrate at 80°C. The temperature was cooled to 70°C and crystal precipitated. The mixture was kept at 70°C for 1 h and then 60°C for 1 h. The mixture was further cooled to 0°C and stirred for 2 h. The crystals of lapatinib ditosylate were isolated by filtration and were dried at 40°C under vacuum overnight. Lapatinib ditosylate (2.5 g, 2.70 mmol, 83% yield) with 99.9% HPLC purity was obtained. XRPD analysis (figure 9) indicated that this was Form 2 as disclosed in WO 2009/079541 Al.
Example 6: Preparation of lapatinib ditosylate monohydrate Lapatinib ditosylate (2.0 g, 96.7% HPLC purity, 2.1 mmol) was dissolved in DMSO (5 mL) at 80°C (internal temperature) and the solution was filtered whilst the lapatinib ditosylate was still dissolved. A mixture of MeCN (5 mL, 2.5 P) and water (0.3 mL) was then added dropwise into the filtered solution at 70-80°C (internal temperature). The mixture was cooled at a rate of 10°C/h until 60°C, and was kept at 60°C for 2 h and was then slowly cooled down to 50°C. After being kept at 50°C for 1 h, MeCN (15 mL) was added, and then the mixture was cooled to 20-30°C and stirred at 20-30°C for 2 h. The slurry was filtered, washed with MeCN (6 mL) and the filter cake was dried in vacuo at 60°C for 4 h to give lapatinib ditosylate monohydrate (1.7 g, 99.4A% purity, 1.8 mmol). XRPD analysis (figure 10) indicated that this was the monohydrate crystalline form as disclosed in US 7,157,466 B2.

.........................................................


Example 3
[0029] Under a nitrogen atmosphere, 2 - furaldehyde diethyl acetal 950g, 9000mL of dry tetrahydrofuran and transferred to the flask, the system was cooled to _40 ° C, n-butyl lithium in tetrahydrofuran (3180mL, 2.2mol / L ) was added dropwise to the reaction system to maintain -4 (T-5 (TC stirred for 2.5 ~ 3h, then triisopropyl borate was added dropwise 1536mL, and stirred for Ih at _60 ° C, after the system was allowed to warm to room temperature, 384mL of glacial acetic acid was slowly added dropwise, followed by stirring for 30min, then dropping 156mL water was added to 3780mL of ethanol, 776mL of triethylamine were then added N_ [3_ chloro _4-[(3_ fluorophenyl) methoxy] phenyl] -6 - iodo-4 - quinazolinamine 1124g, 10% palladium on carbon 134g, and the reaction system was heated to reflux temperature, the reaction 14h. temperature was lowered to room temperature, the reaction mixture was filtered, the filter cake was washed with tetrahydrofuran, The filtrates were combined. To the filtrate was added 240g of triethylamine were then added 2 - (methylsulfonyl) ethylamine 390g and 450mL of methanol, and stirred at room temperature lh, then potassium borohydride was added 137.9g, room temperature for 1.5h, then ice under cooling, a 5N aqueous sodium hydroxide was added dropwise 3600mL, stirred at room temperature 15min, standing layered organic phase was separated, the organic phase p-toluenesulfonic acid was added dropwise 2400g / 3600mL of tetrahydrofuran was stirred for 40min, the solid was filtered and the filter cake was washed with tetrahydrofuran, and then recrystallized from methanol and dried in vacuo to obtain pure final two pairs of p-toluenesulfonic acid lapatinib 1185g. yield 70.8%, purity 98.1%. HNMR (DMSO) 2.27 Cs, 6H) , 3.11 (s, 3H), 3.50 (t, 2H), 3.60 (t, 2H), 4.47 (s, 2H), 5.32 (s, 2H), 6.90 (s, lH), 7.1 (d, J = 7.8 Hz, 4H), 7.19 (t, lH), 7.20 (t, lH), 7.22 (d, J = 3.2Hz, 1H) ,7.23-7 .25 (m, 3H),
7.56 (d, J = 8.0Hz, 4H), 7.62 (dd, Jl = 8.7Hz, J2 = 8.0Hz, 1H), 7.87 (s, 1H), 7.91 (d, J = 8.9Hz, 1H), 8.42 ( d, J = 8.7Hz, 1H), 8.93 (s, lH), 9.03 (s, lH), 9.32 (s, 1H), 11.34 (s, 1H).
..................................
PAPER
Beilstein J. Org. Chem. 2013, 9, 2265–2319.
GlaxoSmithKline’s lapatinib (3.38, Tykerb) is a novel dual kinase inhibitor used in the treatment of solid tumors such as those found in breast cancer and contains a quinazoline core structure. It consists of a 2,5-disubstituted furan ring, which is directly linked to the aminoquinazoline unit (Scheme 41). The quinazoline heterocycle was prepared starting from 5-iodoanthranilic acid (3.72) via initial condensation with formamidine acetate (3.73) followed by chlorination using oxalyl chloride or phosphorous oxychloride [101]. Performing a nucleophilic aromatic substitution on the chloride 3.74 with aniline 3.75renders the extended core of lapatinib. This intermediate (3.76) was then coupled with 5-formyl-2-furanoboronic acid (3.77) using standard Suzuki cross-coupling conditions. Finally, a reductive amination of the pendant aldehyde of3.78 with 2-(methylsulfonyl)ethylamine (3.79) furnishes the desired product lapatinib (Scheme 41).
![[1860-5397-9-265-i41]](http://www.beilstein-journals.org/bjoc/content/inline/1860-5397-9-265-i41.png?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 41: Synthesis of lapatinib.
get ref from
……………………………………………..

Guntrip SB, Lackey KE, Cockerill GS, Carter MC, Smith KJ Bicyclic heteroaromatic compounpds as protein tyrosine kinase inhibitors. EP 1047694; WO 9935146.

Quinazoline ditosylate salt compounds (US7157466)

A NOVEL PROCESS FOR THE PREPARATION OF Lapatinib AND ITS PHARMACEUTICALLY ACCEPTABLE SALTS ( WO 2010061400)
.............................................................
Patent
Fresenius Kabi Oncology Ltd.WO 2013080218
Lahiri, Saswata; Gupta, Nitin; Singh, Hemant Kumar; Handa, Vishal; Sanghani, Sunil
6 JUNE 2013, http://www.google.com/patents/WO2013080218A1?cl=en
Lapatinib of Formula-(II), was first disclosed by SmithKline Beecham in US Patent No. 6,727,256.

The process for the preparation of Lapatinib of Formula-(II), disclosed in W099/35146, is given in the Scheme-I. Accordingly, 4-chloro-6-iodo-quinazoline of Formula-(IV), is reacted with 3-chloro-4-(3'-fluoro-benzyloxy)-aniline yielding N-[3- chloro-4-{(3'-fluorobenzyloxy) phenyl} ]-6-iodo-quinazoline of Formula-( l). The compound of the Formula-(l) reacts with 5-(l,3-dioxolan-2-yl)-2-(tributylstannyl)furan to get the compound of Formula-(2) which on deprotection with HC1, removes the 1,3- dioxolan-2-yl protecting group and liberates 5-(4-{3-chloro-4-(3-fluoro- benzyloxy)anilino}-6- quinazolinyl)-furan-2-carbaldehyde of Formula-(3). The compound of the Formula-(3) on reaction with 2-methanesulfonylethylamine, followed by reductive amination using sodium (triacetoxy)borohydride as the reducing agent gives the required compound Lapatinib of Formula-(II) as an organic residue, which is purified by column chromatography and subsequently converted into its hydrochloride salt (5).

Subsequently, US 7, 157,466 also discloses the preparation of Lapatinib and its ditosylate salt, which is given in Scheme-II.
Lapatinib ditosylate has been prepared by reacting the tosylate salt of 5-(4-[3- chloro-4-(3-fluorobenzyloxy)-anilino]-6-quinazolinyl)-furan-2-carbaldehyde of Formula (3) with 2-(methylsulfonyl)ethylamine in the presence of base (diisopropyl- ethylamine) followed by reduction with sodium triacetoxyborohydride to obtain Lapatinib base which is converted to Lapatinib ditosylate anhydrate by adding para- toulenesulfonic acid. Conversion to Lapatinib ditosylate monohydrate is carried out using THF/H20. Intercon vers ion to the anhydrate of the ditosylate salt and back to monohydrate is carried out with methanol and water respectively.

(lla)
WO201 1039759, filed by Natco Pharma also describes a process for the preparation of Lapatinib from 2-amino benzonitrile, as given in scheme-Ill. Firstly, 2- aminobenzonitrile (6) is reacted with iodine monochloride in acetic acid medium to form compound of Formula (7) which is recrystallized from mixture of hexane and toluene. The compound of Formula (1) is reacted with N,N-dimethylformamide dimethy|acetal in an organic solvent such as toluene or xylene to form novel compound of Formula (8). The compound of Formula (7) is then coupled with compound of Formula (8) in presence of acid catalyst such as trifluoroacetic acid, formic acid or acetic acid to form compound of Formula (3). The compound of Formula (3) is the subjected to Suzuki coupling with 5-formyl-2-furyl boronic acid in ethereal solvent in the presence of catalyst selected from palladium (II) acetate, palladium (II) chloride, and palladium on carbon to form aldehyde compound of Formula (4). The compound of Formula (4) is reacted with 2-methanesulphonyl ethylamine or its salt to produce imine compound of Formula (VI) which is reduced with sodium borohydride to form Lapatinib base (II). The crude Lapatinib base is purified by crystallization from organic solvents. The purified Lapatinib base is converted into Lapatinib ditosylate anhydrous by treating Lapatinib base in organic solvent with /7-toluenesulfonic acid monohydrate which is then recrystallized from aqueous alcohol to produce pharmaceutically acceptable Lapatinib ditosylate monohydrate. The process is depicted in Scheme-Ill.
-IH

Lapatinib (II) WO2010017387, filed by Teva relates to Lapatinib intermediates and process for the preparation of Lapatinib base and Lapatinib ditosylate. The application relates to highly pure intermediate of Formula (2), 3-chloro-4-(3-fluorobenzyloxy)aniline which is prepared by reducing a compound of Formula (1), 3-chloro-4-(3- fluorobenzyloxy)nitrobenzene, with iron and ammonium chloride system in the presence of a C1 -C4 alcohol and water at refluxing temperature. The application also relates to highly pure intermediate of Formula (3), N-[3-chloro-4-(3-fluorobenzyloxy)- phenyl]-6-iodoquinazolin-4-amine, which is prepared in one-pot process from compound of Formula (1 ) by reduction using iron and ammonium chloride system in presence of C1 -C4 alcohol and water. The compound of Formula (3) is reacted with 5- formyl-2-furanboronic acid in the presence of a palladium catalyst and a base in a polar organic solvent to obtain Lapatinib aldehyde base, compound of Formula (4). Optionally, Lapatinib aldehyde base is combined with /? oluenesulfonic acid to obtain Lapatinib aldehyde monotosylate, compound of Formula (5). The invention further provides a process for the preparation of Lapatinib base. Lapatinib aldehyde base or its salt is combined with methylsulfonylethylamine or its hydrochloride salt, acetic acid, an inorganic base in an organic solvent and a reducing agent (sodium triacetoxyborohydride) to form Lapatinib base. Lapatinib base is further purified by using organic solvents. Lapatinib base obtained is further converted to Lapatinib ditosylate. The process is depicted in scheme-IV.
Scheme-IV

Example-5
Preparation of Lapatinib Ditosylate
To a stirred mixture of Sodiumtriacetoxyborohydride (0.21 g) in Tetrahydrofuran (THF)(2.4 ml) was added N-(3-Chloro-4-(3-fluorobenzyloxy)phenyl)-6-(5-((2- (methylsulfonyl)ethylimino)- methyl)furan-2-yl)quinazolin-4-amine (0.2 g) in THF. The reaction mixture was stirred for 1 hour at 20-25 °C. Reaction was monitored by TLC and on completion of reaction, aqueous NaQH (0.16 g NaOH to 0.8 g demineralized water) was added. The organic layer was separated and added p- Toluenesulfonic acid (0.42) in THF (0.6 ml) and stirred for 3 hours. The solid was filtered and dried under vacuum at 60-65°C till constant weight.
Weight: 0.15 g
Yield: 46.9 %
Purity by HPLC: 96.16%
MS (ES+) m/z: 581 [M+H]+ & 583 [M+H+2]+
1H NMR (400 MHz; DMSO-d6): 2.28 (s, 6H), 3.14 (s, 3H), 3.44 (t, J=8.0 Hz, 2H), 3.55 (t, J=8.0 Hz, 2H), 4.46 (s, 2H), 5.31 (s, 2H), 6.89 (br s, 1H), 7.10 (d, J=7.2 Hz, 4H), 7.20 (m, 1H), 7.23 (br s, 1H), 7.31- 7.36 (m, 3H), 7.47 (d, J=7.2 Hz, 4H), 7.63 (d, J=8.8 Hz, IH), 7.89 (br s, IH), 7.92 (d, J=8.8 Hz, IH), 8.39 (d, J=8.8 Hz, IH), 8.89 (s, IH), 8.98 (s, IH), 9.28 (s, IH, NH), 11.18 (s, IH, NH).
.................................................
Patent

..................................................
Patent
The free base and HCl salts of the compounds of Formulae (I), (II), (III), and (IV), may be prepared according to the procedures of International Patent Application No. PCT/EP99100048, filed Jan. 8, 1999, and published as WO 99/35146 on Jul. 15, 1999, referred to above. A schematic of such procedures is presented in Scheme A following. The specific page references given are to WO 99/35146. The free base of the compound of formula II is used as an example of the general scheme.


The compound of formula (II), i.e., N-{3-Chloro-4-[(3-fluorobenzyl) oxy]phenyl}-6-[5-({[2-(methanesulphonyl) ethyl]amino}methyl)-2-furyl]-4-quinazolinamine ditosylate has been prepared in two distinct forms, an anhydrate form (Formula II′ in Scheme B) and a monohydrate form (Formula II″ in Scheme B). The relationship of these forms is illustrated in Scheme B below. The anhydrate form of N-{3-Chloro-4-[(3-fluorobenzyl) oxy]phenyl}-6-[5-({[2-(methanesulphonyl)ethyl]amino}methyl)-2-furyl]-4-quinazolinamine ditosylate may be prepared by (a) reacting the tosylate salt of 5-(4-[3-chloro-4-(3-fluorobenzyloxy)-anilino]-6-quinazolinyl)-furan-2-carbaldehyde (formula B in Scheme B) with 2-(methylsulfone)ethylamine in tetrahydrofuran in the presence of diisopropyl-ethylamine followed by (b) the introduction of this solution into to a slurry of sodium triacetoxyborohydride in tetrahydrofuran at room temperature, (c) adding 5N sodium hydroxide to adjust the pH to within a range of 10–11, (d) separating the organic tetrahydrofuran phase, and then (e) adding para-toulenesulfonic acid hydrate to the organic phase to provide the ditosylate anhydrate. Interconversion to the monohydrate and back to the anhydrate of the ditosylate salt compounds of the invention is as depicted in Scheme B. The tosylate salt of 5-(4-[3-chloro-4-(3-fluorobenzyloxy)-anilino]-6-quinazolinyl)-furan-2-carbaldehyde is prepared from the HCl salt of the carbaldehyde (Formula A of Scheme B). Preparation of N-{3-Chloro-4-[(3-fluorobenzyl) oxy]phenyl}-6-[5-({[2-(methanesulphonyl)ethyl]amino}methyl)-2-furyl]-4-quinazolinamine ditosylate and the anhydrate and monohydrate forms thereof are utilized as an example. As recognized by those skilled in the art, other compounds of formula I and anhydrate and hydrate forms thereof may be prepared by similar methods.

Compound A of Scheme B may be prepared by various synthetic strategies, other that the strategy recited in Scheme A above, utilizing the palladium(O) mediated coupling of quinazoline and substituted furan intermediates.
Example 8
Preparation of N-{3-Chloro-4-[(3-fluorobenzyl) oxy]phenyl}-6-[5-({[2-(methanesulphonyl)ethyl]amino}methyl)-2-furyl]-4-quinazolinamine ditosylate anhydrate (Anhydrate Form of Compound of Formula II)
To a 20 L reactor was added 13.3 vol of THF followed by 0.62 wt (2.93 mol) of NaBH(OAc)3. The 20 L reactor was set to maintain contents at 20° C. A second 20 L reactor was charged with 1000 grams, (1.55 mol) of 5-(4-[3-chloro-4-(3-fluorobenzyloxy)-anilino]-6-quinazolinyl)-furan-2-carbaldehyde 4-methyl benzenesulfonate prepared by the procedure of Example 7 and 6.7 vol of THF. To the THF solution of 5-(4-[3-chloro-4-(3-fluorobenzyloxy)-anilino]-6-quinazolinyl)-furan-2-carbaldehyde 4-methylbenzenesulfonate was added 0.325 vol (1.86 mol) diisopropylethylamine followed by 0.32 wt of 2-(methylsulfone)ethylamine, (321 g, 2.6 mol) and 0.15 vol of IPA. After 1 hour, the preformed imine/THF solution was transferred by vacuum to the stirred suspension of NaBH(OAC)3 in the first 20 L reactor over 10 minutes. After 90 minutes, 4 vol of 5N NaOH was added over 40 min via a pump. This solution was allowed to stir for 15 minutes after which the stirrer was switched off and the layers were allowed to separate. The aqueous layer was drained from the bottom of the reactor and the organic layer transferred to the empty 20 L reactor through a teflon-lined stainless steel jacketed transfer hose outfitted with an in-line 0.45 μm filter. To this solution was added a 2 vol THF solution of 4 wt (1180 g, 6.2 mole) of p-toluenesulfonic acid monohydrate over 5 min. A yellowish precipitate was observed to come out of solution and this was allowed to stir at room temperature for 12 hours. The reaction was drained from the bottom of the reactor and filtered through a ceramic filter lined with paper. The yellow filter cake was washed with 1 vol of a 95:5 THF water solution and allowed to air dry overnight. After suctioning dry for 12 hours, the yellow filter cake was transferred to two glass trays and placed in the drying oven (42° C.) under house vacuum (18 in Hg) with a nitrogen bleed. The two glass trays were removed from the oven and allowed to cool to room temperature and sampled accordingly. The isolated yield of N-{3-Chloro-4-[(3-fluorobenzyl) oxy]phenyl}-6-[5-({[2-(methane-sulphonyl)ethyl]amino}methyl)-2-furyl]-4-quinazolinamine ditosylate (anhydrate) was 1264 grams (1.3 wt, 88%; 1443 g Th) and was a yellow solid.
Approximately 50 mg of the product was transferred to a Karl Fisher Volumetric Moisture Apparatus (model DL35, Mettler, Hightstown, N.J.), which was operated according to the manufacturer's instructions. The anhydrate water content was determined to be 0.31%.
Example 10Preparation of N-{3-Chloro-4-[(3-fluorobenzyl) oxy]phenyl}-6-[5-({[2-(methanesulphonyl)ethyl]amino}methyl)-2-furyl]-4-quinazolinamine ditosylate monohydrate (Monohydrate Form of Compound of Formula II)
A 20 L reactor was charged with 1 wt (930 g, 1.0 mol) of N-{3-Chloro-4-[(3-fluorobenzyl) oxy]phenyl}-6-[5-({[2-(methanesulphonyl)ethyl]amino}methyl)-2-furyl]-4-quinazolinamine ditosylate anhydrate prepared using the procedure of Example 8. To this was added 10 volumes of a pre-mixed 8:2 THF:deionized water solution and the reactor was heated to 65° C. Complete dissolution was observed at 50° C. The clear reaction mixture was transferred to another 20 L reactor through a stainless steel jacketed transfer hose that was equipped with an in-line 5.0 μm cartridge filter. The empty 20 L reactor and the filter line were washed with 0.2 vol of the pre-mixed 8:2 THF:deionized water solution. An additional 1 vol of pre-mixed 8:2 THF:deionized water solution was used to wash the material into the reaction mixture. The 20 L reactor was heated to ˜80° C. The reaction temperature was then ramped down to 55° C. over 2 hours and then to 45° C. over 10 hours. After 10 hours, the temperature was adjusted to 25° C. and the reaction mixture allowed to stir at room temperature for 45 minutes. The yellow precipitate was drained from the bottom of the 20 L reactor into a ceramic filter lined with paper. The flow was fast and smooth and the filter rate very good. The yellow filter cake was washed with 0.6 volumes of a pre-mixed 8:2 THF:deionized water solution and the yellow solid was air dried for 4 hours and placed into a glass tray. The glass tray was placed in a vacuum oven under house vacuum (˜18 in Hg) at 60° C. with a nitrogen bleed for 2 days. After removal from the oven, the material was sampled accordingly. The yield was 743 grams (0.8 wt, 80%; 930 g th) as a bright yellow, crystalline solid.
Approximately 50 mg of the product was transferred to a Karl Fisher Volumetric Moisture Apparatus (model DL35, Mettler, Hightstown, N.J.), which was operated according to the manufacturer's instructions. The monohydrate water content was determined to be 1.99%, which is in agreement with the theoretical value of 1.92%.

Literature References:
Reversible dual inhibitor of ErbB1 and ErbB2 tyrosine kinases. Prepn: M. C. Carter et al., WO 9935146(1999 to Glaxo); eidem, US6727256 (2004 to SmithKline Beecham).
Mechanism of action study: W. Xia et al., Oncogene 21, 6255 (2002); and crystal structure in complex with epidermal growth factor receptor (EGFR, ErbB1): E. R. Wood et al., Cancer Res. 64, 6652 (2004).
In vitro antitumor activity in combination with anti-ErbB2 antibodies: W. Xia et al., Oncogene 24, 6213 (2005). Biologic effects on tumor growth: N. L. Spector et al., J. Clin. Oncol. 23, 2502 (2005).
Pharmacokinetics and clinical activity in metastatic carcinomas: H. A. Burris III et al., ibid. 5305.
Review of clinical development: T. E. Kim, J. R. Murren, IDrugs6, 886-893 (2003); H. A. Burris III, Oncologist 9, Suppl. 3, 10-15 (2004).
Lapatinib Ditosylate [USAN]

- Lapatinib ditosylate monohydrate
- Tykerb
- Tyverb
- UNII-G873GX646R
- KS-1300; 388082-78-8
- N-(3-Chloro-4-((3-fluorobenzyl)oxy)phenyl)-6-(5-(((2-(methylsulfonyl)ethyl)amino)methyl)furan-2-yl)quinazolin-4-amine bis(4-methylbenzenesulfonate) monohydrate
Dosages/Routes/Forms
| STRENGTH | FORM/ROUTE | MARKETING STATUS | RLD | TE CODE |
|---|---|---|---|---|
| EQ 250MG BASE | TABLET;ORAL | 1 | 1 |
Approval History
Derivative Type: Ditoluenesulfonate monohydrate
CAS Registry Number: 388082-78-8; 388082-77-7 (anhydrous)
Additional Names: Lapatinib ditosylate
Manufacturers’ Codes: GW-572016F
Molecular Formula: C29H26ClFN4O4S.2C7H8O3S.H2O
Molecular Weight: 943.48
Percent Composition: C 54.74%, H 4.70%, Cl 3.76%, F 2.01%, N 5.94%, O 18.65%, S 10.20%
Properties: Yellow solid.
Therap-Cat: Antineoplastic.
Keywords: Antineoplastic; Tyrosine Kinase Inhibitors.
References
- Burris HA (2004). "Dual kinase inhibition in the treatment of breast cancer: initial experience with the EGFR/ErbB-2 inhibitor lapatinib". Oncologist. 9 Suppl 3: 10–5.doi:10.1634/theoncologist.9-suppl_3-10. PMID 15163842.
- Higa GM & Abraham J (September 2007). "Lapatinib in the treatment of breast cancer". Expert Review of Anticancer Therapy (log in required) (Future Drugs) 7(9): 1183–92. doi:10.1586/14737140.7.9.1183. PMID 17892419.
- Pazdur, Richard (14 January 2011). "FDA Approval for Lapatinib Ditosylate".Womens Health (Lond Engl) (Cancer.gov) 6 (2): 173. doi:10.2217/whe.10.11.PMID 20187722.
- ^ Jump up to:a b c d "GlaxoSmithKline receives marketing authorisation in the EU for Tyverb (lapatinib), the first oral targeted therapy for ErbB2-positive breast cancer" (Press release). GlaxoSmithKline. 2008-06-12. Retrieved 2008-06-21.
- ^ Jump up to:a b c "GlaxoSmithKline Reports Positive New Data On Tykerb (lapatinib) At The 2007 American Society Of Clinical Oncology (ASCO) Annual Meeting" (Press release). Medical News Today. June 4, 2007. Retrieved December 2, 2008.
- "Data Sheet: TYKERB". Medsafe. New Zealand Medicines and Medical Devices Safety Authority. March 12, 2008. Retrieved December 2, 2008.
- Jump up^ Kulkarni, Kaustubh (2 August 2013). "India revokes GSK cancer drug patent in latest Big Pharma blow". Reuters (Mumbai, India: Reuters). Retrieved 2 August 2013.
- Wood, ER, Truesdale, AT, McDonald, OB, Yuan, D, Hassell, A, Dickerson, SH, Ellis, B, Pennisi, C et al. (2004). "A unique structure for epidermal growth factor receptor bound to GW572016 (Lapatinib): relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells". Cancer Research 64 (18): 6652–9. doi:10.1158/0008-5472.CAN-04-1168. PMID 15374980.
- Dr. Angel Rodriguez (April 2008). "New type of drug shrinks primary breast cancer tumors significantly in just six weeks; research provides leads to a new target in cancer treatment – the cancer stem cell".
- Nelson MH, Dolder CR (February 2006). "Lapatinib: a novel dual tyrosine kinase inhibitor with activity in solid tumors". Ann Pharmacother 40 (2): 261–9.doi:10.1345/aph.1G387. PMID 16418322.
- Jump up^ Geyer CE, Forster J, Lindquist D, et al. (December 2006). "Lapatinib plus capecitabine for HER2-positive advanced breast cancer". N. Engl. J. Med. 355 (26): 2733–43.doi:10.1056/NEJMoa064320. PMID 17192538.
- J Burris HA, Hurwitz HI, Dees EC, et al. (August 2005). "Phase I safety, pharmacokinetics, and clinical activity study of lapatinib (GW572016), a reversible dual inhibitor of epidermal growth factor receptor tyrosine kinases, in heavily pretreated patients with metastatic carcinomas". J. Clin. Oncol. 23 (23): 5305–13.doi:10.1200/JCO.2005.16.584. PMID 15955900.
- J NCI Cancer Drug Information. FDA Approval for Lapatinib Ditosylate (Tykerb®). Retrieved 27 January 2014.
- |url=http://www.bioportfolio.com/news/article/1492867/GSK-Tykerb-Tyverb-Phase-III-gastric-cancer-study-fails-to-meet-primary.html
External links
| WO1999035146A1 | Jan 8, 1999 | Jul 15, 1999 | Glaxo Group Ltd | Bicyclic heteroaromatic compounds as protein tyrosine kinase inhibitors |
| WO2010017387A2 | Aug 6, 2009 | Feb 11, 2010 | Teva Pharmaceutical Industries Ltd. | Lapatinib intermediates |
| WO2011039759A1 | Sep 29, 2009 | Apr 7, 2011 | Natco Pharma Limited | A new process for the preparation of lapatinib and its pharmaceutically acceptable salts |
| US6727256 | Jan 8, 1999 | Apr 27, 2004 | Smithkline Beecham Corporation | 4-aminoquinazoline derivatives as anticarcinogenic agents |
| US7157466 | Jun 28, 2001 | Jan 2, 2007 | Smithkline Beecham (Cork) Limited | Quinazoline ditosylate salt compounds |
| WO1998002434A1 * | Jul 11, 1997 | Jan 22, 1998 | Malcolm Clive Carter | Fused heterocyclic compounds as protein tyrosine kinase inhibitors |
| WO2007121279A2 * | Apr 12, 2007 | Oct 25, 2007 | Tona Morgan Gilmer | Cancer treatment method |
......................................
17
QUIZARTINIB
















































WYETH INNOVATOR
PFIZER DEVELOPER

















17
QUIZARTINIB
QUIZARTINIB
1-(5-(tert-Butyl)isoxazol-3-yl)-3-(4-(7-(2-morpholinoethoxy)benzo[d]imidazo[2,1-b]thiazol-2-yl)phenyl)urea
N-(5-tert-butyl-isoxazol-3-yl)-N’-{ 4- [7-(2-morpholin-4-yl-ethoxy)imidazo [2, 1 -b] [ 1 ,3 ]benzothiazol-2-yl]phenyl } urea
FOR acute myeloidLeukemia,
950769-58-1 CAS
| CAS | 950769-58-1 (free base) 1132827-21-4 (2HCl) |
| Formula | C29H32N6O4S |
| MW | 560.7 |
| Synonim | AC220, AC-010220 ASP-2689 |
PATENTS
U.S. Provisional Patent App. No. 60/743,543, filed March 17, 2006, U.S. Patent App. No. 11/724,992, filed March 16, 2007, and U.S. Patent App. Publication No. 2007/0232604, published October 4, 2007,
BioNews TexasAmbit initiates QUANTUM-R Phase 3 clinical trial of quizartinib in FLT3-ITD …News-Medical.net… the treatment of both newly diagnosed and relapsed FLT3-ITD positive and negative AML patients.
Both the U.S. Food and Drug Administration (FDA) and European Commission have granted orphan drug designation to quizartinib for the treatment of AML.AML, High Risk MDS Therapy
see
Quizartinib
Ambit Biosciences
13 MAY 2013
Ambit Biosciences (NASDAQ:AMBI) is a biotech company that focuses on treatments that inhibit kinases, which are drivers for diseases such as cancer. Three drugs are in development, with the lead one being quizartinib — a Phase 2B trial treatment for acute myeloid leukemia. However, AMBI’s collaboration agreement with Astellas Pharma is set to expire in September, and if it is not replaced, it could mean a delay in Phase 3 trials for quizartinib. Keep in mind that AMBI generated $23.8 million in collaboration revenues last year.
Quizartinib (AC220) is a small molecule receptor tyrosine kinase inhibitor that is currently under development by Ambit Biosciencesfor the treatment of acute myeloid leukaemia. Its molecular target is FLT3, also known as CD135 which is a proto-oncogene.[1]
AC-220 is an angiogenesis inhibitor that antagonizes several proteins involved in vascularization. It was engineered by Ambit Biosciences using KinomeScan technology to potently target FLT3, KIT, CSF1R/FMS, RET and PDGFR kinases. Ambit is developing oral AC-220 in phase III clinical studies for the treatment of relapsed/refractory acute myeloid leukemia (AML) patients with the FMS-like tyrosine kinase-3 (FLT3)-ITD mutation. Early clinical trials are also ongoing for the treatment of advanced solid tumors, for the treatment of refractory or relapsed myelodysplasia, in combination with induction and consolidation chemotherapy for previously-untreated de novo acute myeloid leukemia, and as a maintenance therapy of AML following hematopoietic stem cell transplantation (HSCT). In 2009, orphan drug designation was received both in the U.S. and in the EU for the treatment of AML. In 2009, Ambit Biosciences and Astellas Pharma have entered into a worldwide agreement to jointly develop and commercialize the drug candidate for the treatment of cancer and non-oncology indications. This agreement was terminated in 2013.
Flt3 mutations are among the most common mutations in acute myeloid leukaemia due to internal tandem duplication of Flt3. The presence of this mutation is a marker of adverse outcome.
| Quizartinib is a small molecule with potential anticancer activity. Quizartinib is a selective inhibitor of class III receptor tyrosine kinases, including FMS-related tyrosine kinase 3 (FLT3/STK1), stem cell factor receptor (SCFR / KIT), colony-stimulating factor 1 receptor (CSF1R/FMS) and platelet-derived growth factor receptors (PDGFRs .) Able to inhibition of ligand-independent cell proliferation and apoptosis. Mutations in FLT3 are the most frequent genetic alterations in acute myeloid leukemia (AML) and occur in approximately 30% of cases of AML. | |
| Quizartinib представляет собой малую молекулу с потенциальной противораковой активностью. Quizartinib является селективным ингибитором класса III рецепторов тирозин киназ, в том числе FMS-связанных тирозинкиназы 3 (FLT3/STK1), фактор стволовых клеток рецепторов (SCFR / KIT), колониестимулирующий фактор 1 рецепторов (CSF1R/FMS) и тромбоцитарный рецепторов фактора роста (PDGFRs). Способен к торможению лиганд-независимой клеточной пролиферации и апоптоза. Мутации в FLT3 являются наиболее частыми генетическими изменениями в остром миелобластном лейкозе (ОМЛ) и встречаются примерно в 30% случаев ОМЛ. |
Mechanism
Specifically, Quizartinib selectively inhibits class III receptor tyrosine kinases, including FMS-related tyrosine kinase 3 (FLT3/STK1), colony-stimulating factor 1 receptor (CSF1R/FMS), stem cell factor receptor (SCFR/KIT), and platelet derived growth factor receptors (PDGFRs).
Mutations cause constitutive action of Flt3 leading to resulting in inhibition of ligand-independent leukemic cell proliferation and apoptosis.
Clinical trials
It had good results in a phase II clinical trial for refractory AML – particularly in patients who went on to have a stem cell transplant.[2]
………………………..
WO 2007109120 COMPD B1
EXAMPLE 3: PREPARATION OF N-(5-TERT-BUTYL-ISOXAZOL-3-YL)-N’-{4-[7-(2- MORPHOLIN-4-YL-ETHOXY)IMIDAZO[2,1 -B3[1 ,3]BENZOTHIAZOL-2-YL]PHENYL}UREA [Compound B1]
[00426] A. The intermediate 2-amino-1,3-benzothiazol-6-ol was prepared according to a slightly modified literature procedure by Lau and Gompf. J. Org. Chem. 1970, 35, 4103-4108. To a stirred solution of thiourea (7.6 g, 0.10 mol) in a mixture of 200 ml_ ethanol and 9 ml_ concentrated hydrochloric acid was added a solution of 1 ,4-benzoquinone (21.6 g, 0.20 mol) in 400 mL of hot ethanol. The reaction was stirred for 24 hours at room temperature and then concentrated to dryness. The residue was triturated with hot acetonitrile and the resulting solid was filtered and dried.
[00427] The free base was obtained by dissolving the hydrochloride salt in water, neutralizing with sodium acetate, and collecting the solid by filtration. The product (2-amino-1 ,3-benzothiazol-6-ol) was obtained as a dark solid that was pure by LCMS (M+H = 167) and NMR. Yield: 13.0 g (78 %). NMR (DMSOd6) £7.6 (m, 2H ), 6.6 (d, 1H).
[00428] B. To prepare the intermediate 2-(4-nitrophenyl)imidazo[2,1- b][1 ,3]benzothiazoI-7-ol, 2-amino-1 ,3-benzothiazol-6-ol, (20.0 g, 0.12 mol) and 2-bromo-4′-nitroacetophenone (29.3 g, 0.12 mol) were dissolved in 600 mL ethanol and heated to reflux overnight. The solution was then cooled to 00C in an ice-water bath and the product was collected by vacuum filtration. After drying under vacuum with P2O5 , the intermediate (2-(4- nitrophenyl)imidazo[2,1-_D][1,3]benzothiazol-7-ol) was isolated as a yellow solid. Yield: 17.0 g (46 %) NMR (DMSO-CT6) δ 10 (s, 1 H), 8.9 (s, 1H), 8.3 (d, 2H), 8.1 (d, 2H), 7.8 (d, 1 H), 7.4 (s, 1 H), 6.9 (d, 1 H). [00429] C. To make the 7-(2-morpholin-4-yl-ethoxy)-2-(4-nttro- phenyl)imidazo[2,1-£>][1 ,3]benzothiazo!e intermediate: 2-(4- nitrophenyl)imidazo[2,1-jb][1 ,3]benzothiazol-7-ol, (3.00 g, 9.6 mmol) was suspended in 100 mL dry DMF. To this mixture was added potassium carbonate (4.15 g, 30 mmol, 3 eq), chloroethyl morpholine hydrochloride (4.65 g, 25 mmol, 2.5 eq) and optionally tetrabutyl ammonium iodide (7.39 g, 2 mmol). The suspension was then heated to 900C for 5 hours or until complete by LCMS. The mixture was cooled to room temperature, poured into 800 mL water, and allowed to stand for 1 hour. The resulting precipitate was collected by vacuum filtration and dried under vacuum. The intermediate, (7-(2- morpholin-4-yl-ethoxy)-2-(4-nitro-phenyl)imidazo[2,1-jb][1 ,3]benzothiazole) was carried on without further purification. Yield: 3.87 g (95 %) NMR (DMSO-Cf6) δ 8.97 (s, 1 H), 8.30 (d, 2H), 8.0 (d, 2H), 7.9 (d, 1 H), 7.7 (s, 1 H), 7.2 (d, 1 H), 4.1 (t, 2H), 5.6 (m, 4H), 2.7 (t, 2H).
[00430] D. To make the intermediate 7-(2-morpholin-4-yl-ethoxy)-2-(4- amino-phenyl)!midazo[2, 1 -b][1 ,3]benzothiazole: To a suspension of 7-(2- morpholin-4-yl-ethoxy)-2-(4-nitro-phenyl)imidazo[2,1-ib][1 ,3]benzothiazole (3.87g, 9.1 mmol) in 100 ml_ isopropyl alcohol/water (3:1 ) was added ammonium chloride (2.00 g, 36.4 mmol) and iron powder (5.04 g, 90.1 mmol). The suspension was heated to reflux overnight with vigorous stirring, completion of the reaction was confirmed by LCMS. The mixture was filtered through Celite, and the filtercake was washed with hot isopropyl alcohol (150 ml_). The filtrate was concentrated to approximately 1/3 of the original , volume, poured into saturated sodium bicarbonate, and extracted 3 times with dichloromethane. The combined organic phases were dried over MgSO4 and concentrated to give the product as an orange solid containing a small amount (4-6 %) of starting material. (Yield: 2.75 g 54 %). 80% ethanol/water may be used in the place of isopropyl alcohol /water — in which case the reaction is virtually complete after 3.5 hours and oniy traces of starting material are observed in the product obtained. NMR (DMSO-d6) δ 8.4 (s, 1 H), 7.8 (d, 1 H), 7.65 (d, 1 H), 7.5 (d, 2H), 7.1 (d, 1 H), 6.6 (d, 2H), 4.1 (t, 2H)1.3.6 (m, 4H), 2.7 (t, 2H).
[00431] E. A suspension of 7-(2-morpholin-4-yl-ethoxy)-2-(4-amino- phenyl)imidazo[2,1-b][1 ,3]benzothiazole (4.06 g, 10.3 mmol) and 5-tert- butylisoxazole-3-isocyanate (1.994 g, 12 mmol) in toluene was heated at 120 0C overnight. The reaction was quenched by pouring into a mixture of methylene chloride and water containing a little methanol and neutralized with saturated aqueous NaHCO3 solution. The aqueous phase was extracted twice with methylene chloride, the combined organic extracts were dried over MgSO4 and filtered. The filtrate was concentrated to about 20 ml volume and ethyl ether was added resulting in the formation of a solid. The precipitate was collected by filtration, washed with ethyl ether, and dried under vacuum to give the free base. Yield: 2.342 g (41 %) NMR (DMSO-Cf6) £9.6 (br, 1H), 8.9 (br, 1H), 8.61 (s, 1H), 7.86 (d, 1 H), 7.76 (d, 2H), 7.69 (d, 1 H), 7.51 (d, 2H), 7.18 (dd, 1H), 6.52 (s, 1H), 4.16 (t, 2H), 3.59 (t, 4H), 3.36 (overlapping, 4H), 2.72 (t, 2H), 1.30 (s, 9H). NMR (CDCI3) £9.3 (br, 1H), 7.84 (m, 4H), 7.59 (d, 2H), 7.49 (d, 1 H), 7.22 (d, 1 H), 7.03 (dd, 1 H)1 5.88 (s, 1 H), 4.16 (t, 2H), 3.76 (t, 4H), 2.84 (t, 2H), 2.61 (t, 4H), 1.37 (s, 9H).
[00432] F. For the preparation of the hydrochloride salt, N-(5-tert-butyl- isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2, 1 – b][1 ,3]benzothiazol-2-yl]phenyI}urea hydrochloride, the free base was dissolved in a mixture of 20 ml methylene chloride and 1 ml methanol. A solution of 1.0 M HCI in ethyl ether (1.1 eq.) was added dropwise, followed by addition of ethyl ether. The precipitate was collected by filtration or centrifugation and washed with ethyl ether to give the hydrochloride salt. Yield: 2.44 g (98 %) NMR (DMSO-d6) £11-0 (br, 1 H), 9.68 (s, 1H), 9.26 (s, 1H), 8.66 (s, 1 H), 7.93 (d, 1H), 7.78 (m, 3H), 7.53 (d, 2H), 7.26 (dd, 1H), 6.53 (S, 1 H), 4.50 (t, 2H), 3.97 (m, 2H), 3.81 (t, 2H), 3.6 (overlapping, 4H)13.23 (m, 2H)1 1.30 (s, 9H).
[00433] G. Alternatively, Compound B1 may be made by taking the intermediate from Example 4B and reacting it with chloroethyl morpholine hydrochloride under conditions described in Step C. [00434] H . Λ/-(5-tert-butyl-isoxazol-3-yl)-Λ/’-{4-[5-(2-morpholin-4-yl- ethoxy)imidazo[2,1-6][1 ,3]benzothiazol-2-yl]phenyl}urea hydrochloride, a compound having the general formula (I) where R1 is substituted on the 5 position of the tricyclic ring, was prepared in the manner described in Steps A- F but using the cyciization product 2-amino-benzothiazol-4-ol with 2-bromo-4′- nitroacetophenone in Step A. 1H NMR (DMSO-d6) δ 11.6 (br, 1 H)1 9.78 (br, 1H), 9.56 (br, 1 H), 8.64 (s, 1H)1 7.94 (d, 2H), 7.70 (s, 1H)1 7.56 (d, 2H), 7.45 (t, 1 H), 7.33 (d, 1H), 6.54 (s, 1 H), 4.79 (t, 2H), 3.87 (m, 6H), 3.60 (m, 2H), 3.34 (m, 2H)1 1.30 (s, 9H); LC-MS: ESI 561 (M+H)+. [Compound B11] [00435] I. N-(5-tert-butyl-isoxazol-3-yl)-N’-{4-[6-(2-morpholin-4-yl- ethoxy)imidazo[2,1-b][1 ,3]benzothiazol-2-yl]phenyl}urea hydrochloride [Compound B12] was also prepared by first preparing the benzothiazole starting material, 5 methoxy-benzothiazol-2yl~amine: [00436] To prepare the 5-methoxy-benzothiazol-2-ylamine starting material: To a suspension of (3-methoxy-phenyl)-thiourea (1.822g, 10 mmol) in CH2CI2 (20 ml_) at 0 0C was added dropwise a solution of bromine (1.76 g, 11 mmol) in 10 ml of trichloromethane over a period of thirty minutes. The reaction was stirred for 3 hours at room temperature then heated to 3 hours to reflux for one hour. The precipitate was filtered and washed with dichloromethane. The solid was suspended in saturated NaHCOsand extracted with CH2CI2. The extract was dried over MgSO4 and concentrated to give a white solid (1.716 g, 95%).
………………….
WO 2011056939
N-(5-ieri-butyl- isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l-&][l,3]benzothiazol-2- yl]phenyl}urea (I), or a pharmaceutically acceptable salt, solvate, hydrate, or polymorph thereof. N-(5-ieri-Butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l- / ][!, 3]benzo

N- (5-ieri-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l- &][l,3]benzo-thiazol-2-yl]phenyl}urea (I), or a pharmaceutically acceptable salt, solvate, hydrate, or polymorph thereof, comprising any one, two, three, four, five, six, seven of the steps of:
(A) converting 2-amino-6-alkoxybenzothiazole (II), wherein R1 is a suitable phenolic hydroxyl protecting ;

(II) (III)
(B) reacting 2-amino-6-hydroxybenzothiazole (III) with compound (IV), wherein X is a leaving group, to yield 2-(4-nitrophenyl)imidazo[2,l-b]benzothiazol-7-ol (V);
(C) reacting 2-(4-nitrophenyl)imidazo[2,l-b]benzothiazol-7-ol (V) with compound (VI), wherein X2 is a leaving group, to yield 7-(2-morpholin-4-yl-ethoxy)-2-(4- nitrophenyl)imidazo[ -b]benzothiazole (VII);


(D) reducing 7-(2-morpholin-4-yl-ethoxy)-2-(4-nitrophenyl)imidazo[2, 1- bjbenzothiazole (VII) to yield 7-(2-morpholin-4-yl-ethoxy)-2-(4- am

(E) converting 3-amino-5-£er£-butyl isoxazole (IX) to a 5-?er?-butylisoxazol-3- ylcarbamate derivative (X), wherein R2 is optionally substituted aryl, heteroaryl, alkyl, or cycloalkyl;

(IX) (X)
(F) reacting 7-(2-morpholin-4-yl-ethoxy)-2-(4-aminophenyl)imidazo[2,l- bjbenzothiazole (VIII) with a 5-£er£-butylisoxazol-3-ylcarbamate derivative (X) to yield N-(5-ieri-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l- &][l,3]benzo-

(G) converting N-(5-ieri-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl- ethoxy)imidazo[2,l-&][l,3]benzo-thiazol-2-yl]phenyl}urea to an acid addition salt of N- (5-ieri-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l- b] [ 1 ,3]benzo-thiazol-2-yl]phenyl } urea.
[00128] In certain embodiments, provided herein are processes for the preparation of N-(5-ieri-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l- &][l,3]benzo-thiazol-2-yl]phenyl}urea, or a pharmaceutically acceptable salt, solvate, hydrate, or polymorph thereof, as depicted in Scheme 1, wherein R1, R2, X1, and X2 are defined herein elsewhere. In specific embodiments, provided herein are processes for the preparation of N-(5-ieri-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl- ethoxy)imidazo[2,l-&][l,3]benzo-thiazol-2-yl]phenyl}urea (I), or a pharmaceutically acceptable salt, solvate, hydrate, or polymorph thereof, comprising any one, two, three, four, five, six, seven, of the Steps A, B, C, D, E, F, and G, as depicted in Scheme 1.
Scheme 1 :

A. Preparation of 2-amino-6-hydroxybenzothiazole
1. Example A-l[00252] To a 1-L 3-necked round bottom flask fitted with a condenser, heating mantle, and mechanical stirrer was charged aqueous hydrobromic acid (48%, 632 mL, 5.6 mol, 10 equiv). 2-Amino-6-methoxybenzothiazole (100 g, 0.55 mol, 1 equiv) was added to the above flask over 15 minutes. The reaction temperature was raised slowly to reflux (105-110 °C). A clear dark brown colored solution was observed at about 80 °C. The reflux was continued at 105-110 °C for about 4 hr. The progress of the reaction was monitored by HPLC. When 2-amino-6-methoxybenzothiazole was less than 2%, the reaction was substantially complete.
[00253] The reaction mass was cooled to 0-5 °C and at this point precipitation of a solid was observed. The mixture was maintained at 0-5 °C for 0.5 hr and filtered, and the cake was pressed to remove HBr. The wet cake was transferred to a 2-L round bottom flask fitted with a mechanical stirrer. Saturated aqueous sodium bicarbonate solution (-1500 mL) was added slowly at ambient temperature, whereupon considerable frothing was observed. The pH of the solution was found to be about 6.5 to 7. The mixture was stirred for 0.5 hr at ambient temperature and filtered. The filter cake was washed with water (500 mL), dried on the filter and then under vacuum at 30-35 °C for 10-12 hr to give the product 2-amino-6-hydroxybenzothiazole (82 g, 89% yield, HPLC purity = 99%). JH NMR (DMSO-if6, 500 MHz): δ 7.12 (d, 1H), 7.06 (S, 2H, NH2), 7.01 (d, 1H), 6.64 (dd, 1H); MS (m/z) = 167.1 [M+ + 1].
[00254] Table: Summary of Plant Batches

[00255] HPLC chromatographic conditions were as follows: The column used was XTerra RP8, 250 X 4.6 mm, 5μ or equivalent. Mobile Phase A was buffer, prepared by mixing 3.48 g of dipotassium hydrogen phosphate in 1.0 L of water, and adjusting the pH to 9.0 with phosphoric acid. Mobile Phase B was methanol. The flow rate was 1.0 mL/minute. Detection was set at UV 270 nm. The injection volume was 20 μΐ^, and the sample was diluted with a diluent (Mobile Phase A : Mobile Phase B = 70:30). Test solution was prepared by weighing accurately about 25 mg of sample and transferring it into a 100 mL volumetric flask, dissolving with 20-30 mL of diluent, making up the volume to the mark with diluent, and mixing. The HPLC was performed by separately injecting equal volumes of blank and test solution, and recording the chromatogram for all injections. The purity was calculated by area normalization method.
[00256] Table: HPLC Method

2. Example A-2
[00257] 2-Amino-6-methoxybenzothiazole was reacted with hot aqueous HBr at a temperature of >70 °C for about 3 hours and then the clear solution was cooled to ambient temperature overnight. The precipitated solids were collected, dissolved in hot water and the pH was adjusted to between 4.5-5.5. The resultant solids were collected, dried and re-crystallized from isopropanol. Second crop material was collected. The solids were vacuum dried to give 2-amino-6-hydroxybenzothiazole.
[00258] The reaction progress was monitored by thin layer chromatography (TLC). The product was isolated as a white solid, with 99.4% purity (HPLC area %). JH NMR (300 MHz, DMSO-if6) was collected, which conformed to structure.
3. Example A-3
[00259] A 22-L 3-neck round bottom flask was equipped with a mechanical agitator, thermocouple probe, a reflux condenser, and a heating mantle. The flask was charged with hydrobromic acid (14 L, 123.16 mol, 13.10 equiv). Heating was initiated and 2- amino-6-methoxybenzothiazole was added (1.7 kg, 9.4 mol, 1.00 equiv) over 10 minutes with stirring. The heating of the reaction mixture was continued to reflux, and maintained (>107 °C) for approximately 5 hours. The reaction mixture turned into a clear solution between 75 °C and 85 °C. The reaction progress was monitored by TLC until no starting material was observed (A -0.5 mL reaction mixture aliquot was diluted with -0.5 mL water as a clear solution, neutralized with sodium acetate to pH -5 and extracted with 1 mL dichloromethane. The organic layer was spotted: 5%
methanol/dichloromethane; Rf (product) = 0.35; Rf (starting material) = 0.40).
[00260] The reaction mixture was cooled to – 20 °C (overnight). White solids precipitated. The solids were filtered on a polypropylene filter and pressed to remove as much hydrobromic acid from the solids as possible to facilitate the subsequent pH adjustment step. The slightly wet crude product was dissolved in hot (50 °C) water (5 L). The clear solution was filtered to remove any insoluble material present, and the solids were washed with 50 °C water. The filtrate was cooled to 10 °C. The cooled filtrate was neutralized with sodium acetate (- 1.0 kg) to pH 4.5 to 5.5 with vigorous stirring. A thick white solid precipitated. The solids were collected by filtration, and washed with cool (-10 °C) water (2 x 1.0 L) and air dried.
[00261] The wet crude product was slurried in hot (50 °C) isopropanol (3 L) briefly and allowed to stand in a cool room (-5 °C) overnight. The solids were collected by filtration and washed with methyl ferf-butylether (2 x 500 mL). The solids were dried in a vacuum oven overnight (<30 mm Hg) at 30 °C (first crop). Yield: 1068 g (68%), white solid. HPLC: 99.4% (area). JH NMR (300 MHz, DMSO- ) conformed to structure.
[00262] The organic filtrate was collected in a total volume of 1.0 L, cooled to 10 °C. The off-white solids were precipitated and collected by filtration. The solids were dried in a vacuum oven overnight (<30 mm Hg) at 30 °C (second crop). Yield: 497 g (32%), off-white solid. HPLC: 99.8% (area).
[00263] The overall yield combining the first crop and the second crop was 1565 g, (99%).
B. Preparation of 2-(4-nitrophenyl)imidazo[2,l-b]benzothiazol-7-ol

1. Example B-l[00264] A 3-L 3-neck round bottom flask fitted with a condenser, a heating mantle, and a mechanical stirrer was charged with H-butanol (1.5 L), followed by 2-amino-6- hydroxybenzothiazole (75 g, 0.45 mol, 1.0 equiv), 2-bromo-4′-nitroacetophenone (121 g, 0.50 mol, 1.1 equiv), and sodium bicarbonate (41.6 g, 0.50 mol, 1.0 equiv). The reaction temperature was gradually raised to reflux and maintained at reflux (110-115 °C) for 2-3 hr. During the temperature increase, the reaction mass turned into a clear solution and then immediately changed into an orange colored suspension at 65-75 °C. The progress of the reaction was monitored by HPLC analysis every 1 hr (reaction mass sample was submitted to QC). When the level of 2-amino-6-hydroxybenzothiazole was less than 2%, the reaction was substantially complete.
[00265] The reaction mass was slowly cooled to 50-60 °C and then further cooled to 0-5 °C and stirred for 15 min. The precipitated solids were collected by filtration, and dried on the filter. The wet cake was transferred in to a 1-L round bottom flask, and water (600 mL) was added. The suspension was stirred for 0.5 hr and filtered, and the solid was dried on the filter. The wet cake was again taken in to a 1-L round bottom flask and stirred with acetone (200 mL). The slurry was filtered and washed with acetone (2 X 100 mL), and the solid was dried on the filter, unloaded and further dried in a vacuum oven at ambient temperature to give the product 2-(4-nitrophenyl)imidazo[2,l- b]benzothiazol-7-ol (V) (120 g, 85.7% yield, HPLC purity = 98.7%). JH NMR (DMSO- d6, 500 MHz): δ 9.96 (s, 1H, OH), 8.93 (s, 1H), 8.27 (d, 2H), 8.06 (d, 2H), 7.78 (d, 1H), 7.38 (d, 1H), 6.97 (dd, 1H); MS (m/z) = 312 [M+ + 1].
[00266] Table: Summary of Plant Batches

* Input of 2-amino-6-hydroxybenzothiazole (III)
[00267] HPLC chromatographic conditions were as follows: The column used was XTerra RP8, 250 X 4.6 mm, 5μ or equivalent. Mobile Phase A was buffer, prepared by mixing 3.48 g of dipotassium hydrogen phosphate in 1.0 L of water, and adjusting the H to 9.0 with phosphoric acid. Mobile Phase B was methanol. The flow rate was 1.0 mL/minute. Detection was set at UV 235 nm. The injection volume was 10 μΐ^. The blank was prepared by transferring 200 μΐ. of DMSO and 200 μΐ. of 2M NaOH into a 10 mL volumetric flask, making up the volume to the mark with methanol, and mixing. The test solution was prepared by weighing accurately about 10 mg of sample and transferring it into a 50 mL volumetric flask, dissolving with 1 mL of DMSO and 1 mL of 2M NaOH, sonicating to dissolve, making up the volume to the mark with methanol, and mixing. The HPLC was performed by separately injecting equal volumes of blank and test solution, and recording the chromatogram for all injections. The purity was calculated by area normalization method.
[00268] Table: HPLC Method

2. Example B-2
[00269] A 50-L 3-neck round bottom flask was equipped with a mechanical agitator, a thermocouple probe, a reflux condenser, and a heating mantle. The flask was charged with 2-amino-6-hydroxybenzothiazole (1068 g, 6.43 mol, 1.0 equiv) and ethanol (200 proof, 32.0 L), and the suspension was stirred for 10 minutes. 2-Bromo-4- nitroacetophenone (1667 g, 6.49 mol, 1.01 equiv) was added in one portion. The reaction mixture was heated to reflux (78 °C). The reflux was maintained for approximately 25 hours, resulting in a yellow suspension. The reaction progress was monitored by TLC (20% methanol/ethyl acetate; Rf (product) = 0.85; Rf (starting material) = 0.30). TLC indicated -50% 2-amino-6-hydroxybenzothiazole after 24 hours of reflux. Tetrabutylammonium iodide (10 g) was added and reflux was maintained for an additional 12 hours. TLC indicated -50% starting material still present. Coupling was seen to occur at both the thiazole and the amine.
[00270] The reaction mixture was cooled to 0-5 °C. The solids were collected by filtration, and the yellow solid was washed with ethanol (200 proof, 2 X 1.0 L) and diethyl ether (2 X 1.5 L). The solids were dried in a vacuum oven (<10 mm Hg) at 40 °C. Yield: 930 g (46%), yellow solid. HPLC: 99.5% (area). JH NMR (300 MHz, DMSO-i¾) conformed to structure.
3. Example B-3
[00271] The reaction of Step B was carried out on multiple runs, varying solvents, temperature, and base. The results were summarized in the table below. The product (V) was isolated as yellow or green solids, with 1H NMR consistent with the structure and a purity of greater than about 98% by HPLC analysis.
[00272] Table: Reaction Condition Screening

TBAI = Tetrabutylammonium Iodide
C. Preparation of 7-(2-morpholin-4-yl-ethoxy)-2-(4- nitrophenyl)imidazo[2, 1 -bjbenzothiazole
1. Example C-l
[00273] To a 2000-L glass-lined (GL) reactor was charged DMF (298 kg), and the agitator was started. Under a nitrogen blanket, the reactor was charged with 2-(4- nitrophenyl)imidazo[2,l-&]benzothiazol-7-ol (36.8 kg, 118.2 mol, 1.0 equiv), 4-(2- chloroethyl)morpholine hydrochloride (57.2-66.0 kg, 307.3-354.6 mol, 2.6-3.0 equiv), tetrabutylammonium iodide (8.7 kg, 23.6 mol, 0.2 equiv) and potassium carbonate (49.0 kg, 354.6 mol, 3.0 equiv). The resulting yellow suspension was heated and stirred at 90 + 5 °C for at least 15 minutes, then the temperature was increased to 110 + 5 °C. The mixture was stirred for at least 1 hour and then sampled. The reaction was deemed complete if 2-(4-nitrophenyl) imidazo[2,l-&]benzothiazol-7-ol was <1% by HPLC. If the reaction was not complete, the heating was continued and the reaction sampled. If the reaction was incomplete after 6 hours, additional 4-(2-chloroethyl)morpholine hydrochloride may be charged. In general, additional charges of 4-(2- chloroethyl)morpholine hydrochloride had not been necessary at the given scale.
[00274] The reactor was cooled to 20 + 5 °C and charged with water (247 kg) and acetone (492 kg). The mixture was agitated at 20 + 5 °C for at least 1 hour. The product (VII) was isolated by filtration or centrifuge, and washed with water and acetone, and then dried in a vacuum oven at 45 °C to constant weight to give a yellow solid (46.2 kg, 92% yield, HPLC purity = 97.4% by area). JH NMR (300 MHz, DMSO- ) conformed to structure.
2. Example C-2
[00275] 2-(4-Nitrophenyl)imidazo[2, l-b]benzothiazol-7-ol, 4-(2-chloroethyl)- morpholine hydrochloride, potassium carbonate, and tetrabutylammonium iodide were added to N,N-dimethylformamide forming a yellow suspension that was heated at a temperature of >50 °C for over 3 hours. The reaction was cooled and the solids were collected, slurried into water, filtered, slurried into acetone, filtered and washed with acetone to give yellow solids that were dried under vacuum to give 7-(2-morpholin-4-yl- ethoxy)-2-(4-nitrophenyl)imidazo[2,l-b]benzothiazole.
[00276] The reaction progress was monitored by thin layer chromatography (TLC). The product was isolated as a yellow solid, with 99% purity (HPLC area %), and a water content of 0.20%. Infrared (IR) spectrum was collected, which conformed to structure.
3. Example C-3
[00277] A 50-L 3-neck round bottom flask was equipped with a mechanical agitator, a thermocouple probe, a drying tube, a reflux condenser, and a heating mantle. The flask was charged with 2-(4-nitrophenyl)imidazo [2,l-&]benzothiazol-7-ol (1.770 kg, 5.69 mol, 1.0 equiv), N,N-dimethylformamide (18.0 L), 4-(2-chloroethyl)morpholine hydrochloride (2.751 kg, 14.78 mol, 2.6 equiv), potassium carbonate (2.360 kg, 17.10 mol, 3.0 equiv), and tetrabutylammonium iodide (0.130 kg, 0.36 mol, 0.06 equiv) with stirring. The resulting yellow suspension was heated to about 90 °C to 95 °C, maintaining the temperature for approximately 5 hours. The reaction was monitored by TLC until no starting material was observed (20% methanol / ethyl acetate; Rf (product) = 0.15; Rf (starting material) = 0.85).
[00278] The reaction mixture was cooled to -10 °C, and the yellow solids were collected by filtration on a polypropylene filter pad. The solids were slurried in water (2 X 5 L) and filtered. The crude wet product was slurried in acetone (5 L), filtered, and the solids were rinsed with acetone (2 X 1.5 L). The solids were dried in a vacuum oven (<10 mm Hg) at 45 °C. Yield: 2.300 kg (95%), yellow solid. TLC: R/ = 0.15 (20% methanol / EtOAc). HPLC: 95.7% (area). JH NMR (300 MHz, DMSO-i¾) conformed to the structure.
[00279] Table: Yields from multiple batch runs

4. Example C-4
[00280] To a reactor were added 2-(4-nitrophenyl)imidazo [2,l-&]benzothiazol-7-ol (1.0 kg), 4-(2-chloroethyl)morpholine hydrochloride (1.6 kg), tetrabutylammonium iodide (0.24 kg), and potassium carbonate (1.3 kg, anhydrous, extra fine, hydroscopic). N,N-Dimethylformamide (DMF) (8.6 L) was added into the reactor. The DMF used had water content of no more than 0.05% w/w. The mixture was stirred for between 15 and 30 minutes to render a homogeneous suspension, which was heated to between 85 °C and 95 °C and stirred at between 85 °C and 95 °C for 15 to 30 minutes. The mixture was then heated to between 105 °C and 120 °C and stirred at between 105 °C and 120 °C (e.g. , 115 °C) until the reaction was complete (as determined by HPLC to monitor the consumption of starting material). In some embodiments, if necessary (e.g. , if after 6 hours the reaction was not complete as indicated by HPLC analysis), additional 4-(2- chloroethyl)morpholine hydrochloride (0.03 kg) may be added and the reaction mixture stirred at between 105 °C and 120 °C (e.g. , 115 °C) until reaction completion.
[00281] The reaction mixture was cooled to between 20 °C and 30 °C (e.g. , over a period of 3 hours). To another reactor was added deionized water (7.6 L) and acetone (15 L). The mixture of water and acetone was then added into the reaction mixture while maintaining the temperature at between 20 °C and 30 °C. The mixture was then stirred for 1 to 2 hours at a temperature of between 20 °C and 30 °C. The mixture was filtered, and the solid was washed with deionized water (e.g. , about 45x deionized water) until pH of washes was below 8. The solid was then washed with acetone (9.7 L). The solid was dried under vacuum at a temperature of less than 50 °C until the water content by Karl-Fischer was less than 0.30% w/w and TGA curve showed a mass loss of less than 0.30% w/w at about 229 °C (sampling approximately every 6 hours). The desired product was obtained in about 89% yield having about 99% purity by HPLC.
5. Example C-5
[00282] To a reactor were added 2-(4-nitrophenyl)imidazo [2, l-&]benzothiazol-7-ol (1.0 kg), 4-(2-chloroethyl)morpholine hydrochloride (1.6 kg), and potassium carbonate (1.3 kg, anhydrous, extra fine, hydroscopic). N,N-Dimethylformamide (DMF) (8.6 L) was added into the reactor. The DMF used had water content of no more than 0.05% w/w. The mixture was stirred for between 15 and 30 minutes to render a homogeneous suspension, which was heated to between 95 °C and 120 °C (e.g. , between 100 °C and 105 °C) and stirred at between 95 °C and 120 °C (e.g. , 105 °C) until the reaction was complete (as determined by HPLC to monitor the consumption of starting material). In some embodiments, if necessary (e.g. , if after 6 hours the reaction was not complete as indicated by HPLC analysis), additional 4-(2-chloroethyl)morpholine hydrochloride (0.03 kg) and potassium carbonate (0.024 kg) may be added and the reaction mixture stirred at between 100 °C and 120 °C (e.g. , 105 °C) until reaction completion.
[00283] The reaction mixture was cooled to between 60 °C and 70 °C over a period of at least 60 minutes. Industrial water (6 L) was added to the reactor. The reaction mixture was cooled to between 20 °C and 30 °C. Acetone (6 L) was added to the reactor. The mixture was stirred for 1 to 2 hours at a temperature of between 20 °C and 30 °C. The mixture was filtered, and the solid was washed with industrial water (e.g. , about 45 x industrial water) until pH of washes was below 8. The solid was then washed with acetone (9.7 L). The solid was dried under vacuum at a temperature of less than 50 °C, until the water content by Karl-Fischer was less than 0.30% w/w and TGA curve showed a mass loss of less than 0.30% w/w at about 229 °C (sampling approximately every 6 hours).
6. Example C-6
[00284] To a reactor is added 2-(4-nitrophenyl)imidazo [2, l-&]benzothiazol-7-ol (1.0 kg), 4-(2-chloroethyl)morpholine hydrochloride (1.6 kg), and potassium carbonate (1.3 kg, anhydrous, extra fine, hydroscopic). N,N-Dimethylformamide (DMF) (8.6 L) is added into the reactor. The DMF has a water content of no more than 0.05% w/w. The mixture is stirred for between 15 and 30 minutes to render a homogeneous suspension, which is heated to between 95 °C and 120 °C (e.g. , between 100 °C and 105 °C) and stirred at between 95 °C and 120 °C (e.g. , 105 °C) until the reaction is complete (as determined by HPLC to monitor the consumption of starting material). In some embodiments, if necessary (e.g. , if after 6 hours the reaction is not complete as indicated by HPLC analysis), additional 4-(2-chloroethyl)morpholine hydrochloride (0.03 kg) and potassium carbonate (0.024 kg) may be added and the reaction mixture stirred at between 100 °C and 120 °C (e.g. , 105 °C) until reaction completion.
[00285] The reaction mixture is cooled to between 20 °C and 30 °C (e.g. , over a period of 3 hours). To another reactor is added deionized water (7.6 L) and acetone (15 L). The mixture of water and acetone is then added into the reaction mixture while maintaining the temperature at between 20 °C and 30 °C. The mixture is then stirred for 1 to 2 hours at a temperature of between 20 °C and 30 °C. The mixture is filtered, and the solid is washed with deionized water (e.g. , about 45x deionized water) until pH of washes is below 8. The solid is then washed with acetone (9.7 L). The solid is dried under vacuum at a temperature of less than 50 °C until the water content by Karl-Fischer is less than 0.30% w/w and TGA curve shows a mass loss of less than 0.30% w/w at about 229 °C (sampling approximately every 6 hours). D. Preparation of 7-(2-morpholin-4-yl-ethoxy)-2-(4- aminophenyl)imidazo [2, 1 -bjbenzothiazole

[00286] To a 200-L high pressure (HP) reactor was charged a slurry of 7-(2- morpholin-4-yl-ethoxy)-2-(4-nitrophenyl)imidazo [2,l-&]benzothiazole (VII) (7.50 kg, 17.7 mol, 1.0 equiv) in methanol (30 kg). The container was rinsed with additional methanol (10 kg) and the rinse was charged to the reactor. The reactor was then charged with THF (67 kg) and methanol (19 kg). The contents were agitated and the reactor was flushed with nitrogen by alternating nitrogen and vacuum. Vacuum was applied to the reactor and Raney Ni catalyst (1.65 kg, 0.18 wt. equiv) was charged through a sample line. Water (1 kg) was charged through the sample line to rinse the line. The reactor was flushed with nitrogen by alternating nitrogen and vacuum. The reactor was then vented and heated to 50 °C. The reactor was closed and pressurized with hydrogen gas to 15 psi keeping the internal temperature below 55 °C. The reactor was vented and re- pressurized a total of 5 times, then pressurized to 150 psi with hydrogen gas. The contents were agitated at 50 °C for at least 4 hours. At this point a hydrogen uptake test was applied: The reactor was re-pressurized to 150 psi and checked after 1 hour. If a pressure drop of more than 5 psi was observed, the process was repeated. Once the pressure drop remained < 5 psi, the reactor was vented and sampled. The reaction was deemed complete when 7-(2-morpholin-4-yl-ethoxy)-2-(4-nitrophenyl)imidazo [2,1- 6]benzothiazole (VII) was < 0.5% by HPLC.
[00287] The reactor was flushed with nitrogen as shown above. The 200-L HP reactor was connected to the 2000-L GL reactor passing through a bag filter and polish filter. The bag filter and polish filter were heated with steam. The 200-L HP reactor was pressurized (3 psi nitrogen) and its contents were filtered into the 2000-L reactor. The filtrates were hot. The 200-L reactor was vented and charged with THF (67 kg) and methanol (59 kg), the reactor agitated, and filtered into the 2000-L GL reactor.
[00288] A total of 6 reductions (46.2 kg processed) were carried out and the combined batches were concentrated by vacuum distillation (without exceeding an internal temperature of 40 °C) to a volume of -180 L. The reactor was cooled to 20 °C and charged with heptane (250 kg) and again vacuum distilled to a volume of -180 L. The reactor was charged with heptane (314 kg) and agitated at 20 °C for at least 1 hour, and then the product was isolated by centrifugation or collection on a Nutsche filter, washing with heptanes (2-5 kg per portion for centrifugation, followed by a 10-20 kg heptanes rinse of the reactor; or 94 kg for Nutsche filtration, rinsing the reactor first). The cake was blown dry, transferred to a vacuum oven and dried to constant weight maintaining a temperature < 50 °C to give the desired product (VIII) (34.45 kg, 80% yield, HPLC purity = 97.9%).
2. Example D-2
[00289] 7-(2-Morpholin-4-yl-ethoxy)-2-(4-nitrophenyl)imidazo[2,l-b]benzothiazole was dissolved into methanol and THF and placed in a hydrogenator. Raney nickel was added and the vessel was pressurized with hydrogen and stirred for >24 hours. The reaction mixture was concentrated to a thick paste and diluted with methyl ferf-butyl ether. The resulting solids were filtered and washed with methyl ferf-butyl ether and dried under vacuum to give 7-(2-morpholin-4-yl-ethoxy)-2-(4-aminophenyl) imidazo [2, 1 -bjbenzothiazole.
[00290] The reaction progress was monitored by thin layer chromatography (TLC). The product was isolated as a yellow solid, with 99% purity (HPLC area %). IR was collected, which conformed to structure.
3. Example D-3
[00291] Into a 5-gallon autoclave, 7-(2-morpholin-4-yl-ethoxy)-2-(4-nitrophenyl) imidazo[2,l-&]benzothiazole (580 g, 1.37 mol, 1.0 equiv), THF (7.5 L), methanol (7.5 L, AR) and -55 g of decanted Raney nickel catalyst were added. The reaction vessel was purged with nitrogen (3 X 50 psi) and hydrogen (2 X 50 psi), with stirring briefly under pressure and then venting. The autoclave was pressurized with hydrogen (150 psi). The mixture was stirred and the hydrogen pressure was maintained at 150 psi for over 24 hours with repressurization as necessary. The reaction progress was monitored by TLC (10% methanol / chloroform with 1 drop ammonium hydroxide; Rf (product) 0.20; Rf (SM) 0.80). The reaction was substantially complete when the TLC indicated no starting material present, typically after 24 hours of stirring at 150 psi. The hydrogenation was continued at 150 psi for a minimum of 4 hours or until completion if starting material was still present after the initial 4 hours.
[00292] The reaction mixture was filtered through a Buchner funnel equipped with a glass fiber filter topped with a paper filter. Unreacted starting material was removed together with the catalyst. The filtrate was concentrated to a total volume of 0.5 L, and the residue was triturated with methyl ferf-butyl ether (0.5 L). The resultant solids were collected by filtration, and washed with methyl ferf-butyl ether (0.3 L) (first crop).
[00293] The filtrate was concentrated to dryness and the residue was diluted with methyl ferf-butyl ether (2 L). The resultant solids were collected by filtration, washing with methyl ferf-butyl ether (0.5 L) (second crop).
[00294] The solids were dried in a vacuum oven (<10 mm Hg) at 25 °C. Yield: 510 g (95%), beige solid. TLC: R/ 0.2 (10% methanol / chloroform with one drop of ammonium hydroxide). HPLC: 99.0% (area). JH NMR (300 MHz, DMSO-i¾) conformed to the structure.
[00295] Table: Yields from multiple batch runs

4. Example D-4
[00296] The reaction of Step D was carried out in multiple runs under various conditions, such as, e.g. , varying catalyst loading, concentration of reactant, reaction temperature, and/or workup procedures. The results are summarized in the table below.

Description Run # l Run # 2 Run # 3 Run # 4 Run # 5Rxn Temp (°C) RT RT RT RT RT
Rxn Time (Hr) 24 hr 24 hr 24 hr 24 hr 24 hr
Filtered the Filtered the solution
Filtered the Filtered the Filtered the
solution through through celite. The solution through solution through solution through
celite, washed celite filter cake celite, celite, celite,
with THF, refluxed in THF concentrated, concentrated, concentrated,
concentrated, washed with hot solvent exchanged solvent exchanged solvent exchanged
Work Up solvent exchanged THF, concentrated, with heptane, with heptane, with heptane,
with heptane, solvent exchanged stirred the solids stirred the solids stirred the solids
stirred the solids with heptane, stirred and filtered and filtered and filtered
and filtered the solids and washed with washed with washed with
washed with filtered washed with heptane heptane heptane
heptane heptane
Produce (VIII) 1.9 g 3.88 g 1.11 g 2.6 g 4.4 g
Yield 88% 83.4% 56 94.6%
HPLC purity 95.6% 77.5% 91% 93.8%

5. Example D-5
[00297] To a pressure reactor under nitrogen atmosphere was added a slurry of Raney Nickel in water (0.22 kg) (e.g. about 0.14 kg dry catalyst in water) and the charging line was rinsed with deionized water (0.13 L). To another reactor (Reactor B) were added methanol (5.05 L) and 7-(2-morpholin-4-yl-ethoxy)-2-(4-nitrophenyl)imidazo [2, 1- &]benzothiazole (1.0 kg), and the mixture was stirred for between 15 and 30 minutes to render a homogenous suspension. The suspension was transferred to the pressure reactor. Reactor B was washed with methanol (4.88 L) and the wash was transferred to the pressure reactor. Tetrahydrofuran (10.1 L) was added to the pressure reactor.
Hydrogen was charged to the pressure reactor to a pressure of between 2.0 bar and 3.0 bar. The reactor was heated to a temperature of between 45 °C and 55 °C. Hydrogen was then charged to the pressure reactor to a pressure of between 6.0 bar and 7.0 bar. The mixture was stirred at a temperature of between 45 °C and 55 °C (e.g. , 50 °C), while maintaining the hydrogen pressure between 6.0 bar and 7.0 bar until reaction completion (as determined by HPLC to monitor the consumption of starting material).
[00298] The mixture was filtered while maintaining the temperature at between 35 °C and 50 °C. The pressure reactor and the filter were washed with a mixture of THF (10.1 L) and methanol (9.93 L) preheated to a temperature of between 45 °C and 55 °C (e.g. , 50 °C). The combined filtrate was concentrated to a volume of between 2.4 L and 2.8 L under vacuum at a temperature of no more than 40 °C, and a precipitate was formed. Methanol (7.5 L) was added. The slurry was cooled to a temperature of between 5 °C and -5 °C (e.g. , over 3 hours) and stirred for at least 1 hour (e.g. , for 3 hours) while maintaining the temperature at between 5 °C and -5 °C. The suspension was filtered. The solid was washed with methanol (2 X 1.2 L). The solid was then dried under vacuum at a temperature of less than 50 °C until the methanol and THF contents were each less than 3000 ppm as analyzed by GC (e.g. , less than 1500 ppm). The desired product was obtained in about 88.5% yield having about 99% purity by HPLC.
E. Preparation of phenyl 5-£er£-butylisoxazol-3-ylcarbamate

[00299] The jacket temperature of a 200-L glass-lined (GL) reactor was set to 20 °C. To the reactor was charged 5-ieri-butylisoxazole-3-amine (15.0 kg, 107.0 mol, 1.0 equiv), then K2C03 (19.5 kg, 141.2 mol, 1.3 equiv) and anhydrous THF (62 kg).
Agitation was started and then phenyl chloroformate (17.6 kg, 112.4 mol, 1.05 equiv) was charged. The charging line was rinsed with additional anhydrous THF (5 kg). The reaction was agitated at 20 + 5 °C for at least 3 hours then sampled. The reaction was deemed complete if 5-£er£-butylisoxazole-3-amine was < 2% by HPLC. If the reaction was not complete after 6 hours, additional K2CO3 and phenyl chloroformate may be added to drive the reaction to completion.
[00300] Once complete, the reaction was filtered (Nutsche). The filter was rinsed with THF (80 kg). The filtrate was vacuum distilled without exceeding an internal temperature of 40 °C until -50 L remained. Water (188 kg) and ethanol (45 L) were charged, and the mixture was agitated for at least 3 hours with a jacket temperature of 20 °C. The resulting solid was isolated by centrifugation or collection on a Nutsche filter, rinsed with water (2-5 kg for each centrifugation portion; 30 kg for Nutsche filtration) and blow-dried. The solid was then dried to constant weight in a vacuum oven (45 °C) to give the desired product (19.4 kg, 92% yield, HPLC purity = 97.4%). On an 800 g scale, 1559 g of the desired product (98% yield) was obtained with a 99.9% HPLC purity. JH NMR (DMSO-i¾) δ 11.17 (s, 1H); 7.4 (m, 2H); 7.2 (m, 3H); 1.2 (s, 9H). LCMS (M+H)+ 261.
F. Preparation of N-(5-ieri-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl- ethoxy)imidazo[2, 1 -b] [ 1 ,3 ]benzothiazol-2-yl]phenyl } urea

1. Example F-l
[00301] The jacket of a 2000-L GL reactor was set to 20 °C and the reactor was charged with 7-(2-morpholin-4-yl-ethoxy)-2-(4-aminophenyl)imidazo[2,l- &]benzothiazole (26.7 kg, 67.8 mol, 1.0 equiv), 3-amino-5-?-butylisoxazole phenyl carbamate (19.4 kg, 74.5 mol, 1.1 equiv), DMAP (0.5 kg, 4.4 mol, 0.06 equiv), and DCM (anhydrous, 260 kg). Agitation was started, triethylamine (1.0 kg, 10.2 mol, 0.15 equiv) was charged followed by additional DCM (5 kg) through the charging line. The reaction was heated to reflux (-40 °C) and agitated for at least 20 hours with complete dissolution observed followed by product crystallizing from solution after -30 minutes. The reaction was sampled and deemed complete when HPLC analysis showed a ratio of compound (VIII) to compound (I) < 1%.
[00302] The reactor was cooled to 0 °C and stirred for at least 2 hours. The content of the reactor were isolated by centrifuge. Each portion was rinsed with 2-3 kg of cold (0 °C) DCM and spun dry for at least 5 minutes with a 10 psi nitrogen purge. For the final portion, the reactor was rinsed with 10 kg of cold (0 °C) DCM and transferred to the centrifuge where it was spun dry for at least 5 minutes with a 10 psi nitrogen purge. The combined filter cakes were transferred to a vacuum tray dryer and dried to constant weight at 50 °C and at least >20 inches of Hg to give the desired product (I) (35.05 kg, 92% yield, HPLC purity = 98.8%). Phenol was the major impurity detected (0.99%); and three other impurities (<0.10%) were detected. JH NMR (300 MHz, DMSO- ) conformed to structure.
2. Example F-2
[00303] A variety of solvents were used in the reaction of Step F to optimize for better yields and purity profiles. The contents of the symmetrical urea impurity (XI) were compared and summarized in the table below:

http://www.google.com/patents/WO2011056939A1?cl=en SE THIS FOR DELETED CLIPS


4. Example F-4
[00305] To Reactor A under a nitrogen atmosphere was added 7-(2-morpholin-4-yl- ethoxy)-2-(4-aminophenyl)imidazo[2,l-&]benzothiazole (1 kg) and DMAP (0.02 kg). To Reactor B under a nitrogen atmosphere was added 3-amino-5-?-butylisoxazole phenyl carbamate (0.73 kg) and DCM (5.6 L). The DCM used had a water content of less than 0.05 % w/w. The mixture in Reactor B was stirred until dissolution. The solution was transferred into Reactor A (the solution can be filtered into Reactor A to remove any insoluble impurities in the carbamate starting material), and the mixture was stirred in Reactor A. Reactor B was washed with DCM (0.8 L) and the wash was transferred into Reactor A. Reactor A was washed with DCM (0.9 L). To Reactor A was added triethylamine (0.1 L) and the charging vessel and lines were rinsed with DCM (0.1 L) into Reactor A. The mixture was then heated to reflux and stirred at reflux until reaction completion (as determined by HPLC to monitor the consumption of starting material).
[00306] The reaction mixture was cooled (e.g. , over 2 to 3 hours) to a temperature of between -5 °C and 5 °C (e.g. , 0 °C). The mixture was then stirred for 2 to 3 hours at a temperature of between -5 °C and 5 °C (e.g. , 0 °C). The suspension was filtered. The solid was washed with cool DCM (2 X 1.5 L) (pre-cooled to a temperature of between -5 °C and 5 °C). The solid was dried under vacuum at a temperature of less than 45 °C until the DCM content was less than 1000 ppm (e.g., below 600 ppm) as analyzed by GC. The desired product was obtained having about 99% purity by HPLC.
G. Preparation of N-(5-ieri-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl- ethoxy)imidazo[2, l-b] [1 ,3]benzothiazol-2-yl]phenyl }urea dihydrochloride

1. Example G-l
[00307] The jacket of a 2000-L GL reactor was set to 20 °C and the reactor was charged with N-(5-ieri-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo [2, 1-&][1, 3]benzothiazol-2-yl]phenyl}urea (35.0 kg, 62.4 mol, 1.0 equiv) followed by methanol (553 kg). Agitation was started and the reaction mixture was heated to reflux (-65 °C). Concentrated aqueous HC1 (15.4 kg, 156.0 mol, 2.5 equiv) was charged rapidly (<5 minutes) and the charge line was rinsed into the reactor with methanol (12 kg). Addition of less than 2.0 equivalents of HC1 normally resulted in the formation of an insoluble solid. The reaction mixture was heated at reflux for at least 1 hour. Upon HC1 addition, the slurry dissolved and almost immediately the salt started to crystallize, leaving insufficient time for a polish filtration.
[00308] The reactor was cooled to 20 °C and the product was isolated by filtration (Nutsche) rinsing the reactor and then the cake with methanol (58 kg). The solid was then dried in a vacuum oven (50 °C) to constant weight to give the desired
dihydrochloride salt (35 kg, 89% yield, HPLC purity = 99.94%). JH NMR (300 MHz, DMSO-i¾) conformed to structure.
2. Example G-2
[00309] Concentrated HC1 was added to a suspension of N-(5-ieri-butyl-isoxazol-3- yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l-&][l,3]benzothiazol-2- yl]phenyl}urea in warm methanol forming a solution that slowly began to precipitate. The reaction mixture was refluxed for over 2 hours and then stirred overnight at ambient temperature. The dihydrochloride salt was collected and dried under vacuum.
3. Example G-3
[00310] A 50-L 3-neck round bottom flask was equipped with a mechanical agitator, a thermocouple probe, a nitrogen inlet, a drying tube, a reflux condenser, an addition funnel, and a heating mantle. The flask was charged with N-(5-ieri-butyl-isoxazol-3-yl)- N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l-&][l,3]benzothiazol-2-yl]phenyl}urea (775 g, 1.38 mol, 1.0 equiv) and MeOH (40 L, AR). The resulting off-white suspension was heated to reflux (68 °C). A clear solution did not form. HC1 (37% aqueous) (228 mL, 3.46 mol, 2.5 equiv) was added over 5 minutes at 68 °C. The reaction mixture turned into a clear solution and then a new precipitate formed within approximately 3 minutes. Heating was continued at reflux for approximately 5 hours. The reaction mixture was allowed to cool to ambient temperature overnight. The off-white solids were collected by filtration on a polypropylene filter, washing with MeOH (2 X 1 L, AR). [00311] Two lots of material prepared in this manner were combined (740 g and 820 g). The combined solids were slurried in methanol (30 L) over 30 minutes at reflux and allowed to cool to the room temperature. The solids were collected by filtration on a polypropylene filter, rinsing with methanol (2 X 1.5 L). The solids were dried in a vacuum oven (<10 mm Hg) at 40 °C. Yield: 1598 g (84%), off-white solid. HPLC: 98.2% (area). MS: 561.2 (M+l)+. JH NMR (300 MHz, DMSO-i¾) conformed to the structure. Elemental Analysis (EA): Theory, 54.97 %C; 5.41 %H; 13.26 %N; 5.06 %S; 11.19 %C1; Actual, 54.45 %C; 5.46 %H; 13.09 %N; 4.99 %S; 10.91 %C1.
4. Example G-4
[00312] Into a 50-L 3-neck round bottom flask equipped with a mechanical stirrer, a heating mantle, a condenser and a nitrogen inlet, were charged N-(5-ieri-butyl-isoxazol- 3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l-&][l,3]benzothiazol-2- yl]phenyl}urea (1052.4 g, 1.877 mol, 1.0 equiv) and methanol (21 L). The reactor was heated and stirred. At an internal temperature of > 50 °C, cone. HC1 (398.63 mL, 4.693 mol, 2.5 equiv) was charged over 5 minutes through an addition funnel. During the addition, the mixture changed from a pale yellow suspension to a white suspension. The internal temperature was 55 °C at the conclusion of the addition. The mixture was heated to reflux for 1 hour, then heating was discontinued and the mixture was allowed to cool to room temperature. The mixture was filtered in two portions, and each filter cake was washed with methanol (2 X 1 L), transferred to trays and dried in a vacuum oven (45 °C) to constant weight. The dried trays were combined to produce 1141.9 g of the salt (96% yield, 99.1 % HPLC purity, 10.9% chloride by titration).
H. Analytical Data
1. N-(5-ieri-butyl-isoxazol-3-yl)-N’-{ 4-[7-(2-morpholin-4-yl- ethoxy)imidazo[2, l-&] [l ,3]benzothiazol-2-yl]phenyl}urea
dihydrochloride
[00314] A batch of about 30 grams of N-(5-ieri-butyl-isoxazol-3-yl)-N’- {4-[7-(2- morpholin-4-yl-ethoxy)imidazo[2, l-&] [l ,3]benzothiazol-2-yl]phenyl}urea
dihydrochloride was prepared using the methods described herein. This lot was
prepared in accordance with the requirements for production of clinical Active
Pharmaceutical Ingredients (APIs) under GMP conditions. The analytical data for this batch was obtained, and representative data were provided herein. [00315] Summary of analytical data for the dihydrochloride salt.

………………………
EXAMPLE 1. SYNTHESIS OF N-(5-TERT-BUTYL-ISOXAZOL-3-YU- N>-{4-f7-(2-MORPHOLIN-4- YL-ETHOXY)IMID AZO[2,1- BlH,31BENZOTHIAZOL-2-YL|PHENYLiUREA (“COMPOUND Bl”)
[00357] A. The intermediate 2-amino-l,3-benzothiazol-6-ol was prepared according to a slightly modified literature procedure by Lau and Gompf: J. Org. Chem. 1970, 35, 4103- 4108. To a stirred solution of thiourea (7.6 g, 0.10 mol) in a mixture of 200 mL ethanol and 9 mL concentrated hydrochloric acid was added a solution of 1 ,4-benzoquinone (21.6 g, 0.20 mol) in 400 mL of hot ethanol. The reaction was stirred for 24 hours at room temperature and then concentrated to dryness. The residue was triturated with hot acetonitrile and the resulting solid was filtered and dried.
[00358] The free base was obtained by dissolving the hydrochloride salt in water, neutralizing with sodium acetate, and collecting the solid by filtration. The product (2- amino-l,3-benzothiazol-6-ol) was obtained as a dark solid that was pure by LCMS (M+H = 167) and NMR. Yield: 13.0 g (78 %). NMR (DMSO-^) Sl.6 (m, 2H), 6.6 (d, IH). [00359] B. To prepare the 2-(4-nitrophenyl)imidazo[2,l-b][l,3]benzothiazol-7-ol intermediate, 2-amino-l,3-benzothiazol-6-ol (20.0 g, 0.12 mol) and 2-bromo-4′- nitroacetophenone (29.3 g, 0.12 mol) were dissolved in 600 mL ethanol and heated to reflux overnight. The solution was then cooled to O0C in an ice-water bath and the product was collected by vacuum filtration. After drying under vacuum with P2O5 , the intermediate (2- (4-nitrophenyl)imidazo[2,l-£][l,3]benzothiazol-7-ol) was isolated as a yellow solid. Yield: 17.0 g (46 %) NMR (DMSO-(I6) δ 10 (s, IH), 8.9 (s, IH), 8.3 (d, 2H), 8.1 (d, 2H), 7.8 (d, IH), 7.4 (s, IH), 6.9 (d, IH).
[00360] C. To make the 7-(2-morpholin-4-yl-ethoxy)-2-(4-nitro-phenyl)imidazo[2,l-
6][l,3]benzothiazole intermediate: 2-(4-nitrophenyl)imidazo[2,l-6][l,3]benzothiazol-7-ol,
NYI-4144519vl 84 (3.00 g, 9.6 mmol) was suspended in 100 mL dry DMF. To this mixture was added potassium carbonate (4.15 g, 30 mmol, 3 eq), chloroethyl morpholine hydrochloride (4.65 g, 25 mmol, 2.5 eq) and optionally tetrabutyl ammonium iodide (7.39 g, 2 mmol). The suspension was then heated to 900C for 5 hours or until complete by LCMS. The mixture was cooled to room temperature, poured into 800 mL water, and allowed to stand for 1 hour. The resulting precipitate was collected by vacuum filtration and dried under vacuum. The intermediate, (7-(2-morpholin-4-yl-ethoxy)-2-(4-nitro-phenyl)imidazo[2, 1 – b][\, 3]benzothiazole) was carried on without further purification. Yield: 3.87 g (95 %) NMR (DMSO-d6) δ 8.97 (s, IH), 8.30 (d, 2H), 8.0 (d, 2H), 7.9 (d, IH), 7.7 (s, IH), 7.2 (d, IH), 4.1 (t, 2H), 5.6 (m, 4H), 2.7 (t, 2H).
[00361] D. To make the intermediate 7-(2-morpholin-4-yl-ethoxy)-2-(4-amino- phenyl)imidazo[2,l-b][l,3]benzothiazole: To a suspension of 7-(2-morpholin-4-yl-ethoxy)- 2-(4-nitro-phenyl)imidazo[2,l -b][\ , 3]benzothiazole (3.87g, 9.1 mmol) in 100 mL isopropyl alcohol/water (3:1) was added ammonium chloride (2.00 g, 36.4 mmol) and iron powder (5.04 g, 90.1 mmol). The suspension was heated to reflux overnight with vigorous stirring, completion of the reaction was confirmed by LCMS. The mixture was filtered through Celite, and the filtercake was washed with hot isopropyl alcohol (150 mL). The filtrate was concentrated to approximately 1/3 of the original volume, poured into saturated sodium bicarbonate, and extracted 3 times with dichloromethane. The combined organic phases were dried over MgSO4 and concentrated to give the product as an orange solid containing a small amount (4-6 %) of starting material. (Yield: 2.75 g 54 %). 80% ethanol/water may be used in the place of isopropyl alcohol /water – in which case the reaction is virtually complete after 3.5 hours and only traces of starting material are observed in the product obtained. NMR (DMSO-Λfc) δ 8.4 (s, IH), 7.8 (d, IH), 7.65 (d, IH), 7.5 (d, 2H), 7.1 (d, IH), 6.6 (d, 2H), 4.1 (t, 2H), 3.6 (m, 4H), 2.7 (t, 2H).
[00362] E. A suspension of 7-(2-morpholin-4-yl-ethoxy)-2-(4-amino- phenyl)imidazo[2,l-b][l,3]benzothiazole (4.06 g, 10.3 mmol) and 5-tert-butylisoxazole-3- isocyanate (1.994 g, 12 mmol) in toluene was heated at 120 0C overnight. The reaction was quenched by pouring into a mixture of methylene chloride and water containing a little methanol and neutralized with saturated aqueous NaHCO3 solution. The aqueous phase was extracted twice with methylene chloride, the combined organic extracts were dried over
NYI-4144519vl 85 MgSO4 and filtered. The filtrate was concentrated to about 20 ml volume and ethyl ether was added resulting in the formation of a solid. The precipitate was collected by filtration, washed with ethyl ether, and dried under vacuum to give the free base of Compound B 1. Yield: 2.342 g (41 %) NMR (DMSO-J6) £9.6 (br, IH), 8.9 (br, IH), 8.61 (s, IH), 7.86 (d, IH), 7.76 (d, 2H), 7.69 (d, IH), 7.51 (d, 2H), 7.18 (dd, IH), 6.52 (s, IH), 4.16 (t, 2H), 3.59 (t, 4H), 3.36 (overlapping, 4H), 2.72 (t, 2H), 1.30 (s, 9H). NMR (CDCl3) £9.3 (br, IH), 7.84 (m, 4H), 7.59 (d, 2H), 7.49 (d, IH), 7.22 (d, IH), 7.03 (dd, IH), 5.88 (s, IH), 4.16 (t, 2H), 3.76 (t, 4H), 2.84 (t, 2H), 2.61 (t, 4H), 1.37 (s, 9H).
6.2 EXAMPLE 2. ALTERNATIVE SYNTHESIS QF N-(5-TERT-BUTYL- ISOXAZQL-3- YL)-N -{4-[7-q-MORPHOLIN-4- YL- ETHOXYUMID AZOf2,l-BUl,31BENZOTHIAZOL-2- YLIPHENYLIUREA (“COMPOUND Bl”)
[00363] A. To a suspension of the intermediate 2-(4-Nitrophenyl)imidazo[2,l- b][l,3]benzothiazol-7-ol from Example IB (2.24 g, 7.2 mmol) in ethanol (40 mL) was added SnCl2 1H2O (7.9Og, 35 mmol) and heated to reflux. Concentrated HCl was added to the reaction mixture and the precipitate formed gradually. The reaction mixture was heated to reflux for 20 hours and then allowed to cool to room temperature. The solution was poured into ice and neutralized with 10% NaOH and adjusted to approximately pH 6. The organic phase was extracted three times with ethyl acetate (80 mL x 3). Extracts were dried over MgSθ4 and concentrated to give a yellow solid. (1.621 g, 80%). The solid was recrystallized from methanol to give a pure product (1.355 g, 67%).
[00364] B. To a suspension of the intermediate from Step 2A (0.563 g, 2 mmol) in toluene (30 mL) was added 5-tert-butylisoxazole-3-isocyanate (0.332g, 2 mmol) and heated to reflux overnight. LC-MS analysis showed presence of the intermediate but no trace of 5- tert-butylisoxazole-3-isocyanate and an additional 0.166 g of the isocyanate was added. The reaction was again heated to reflux overnight. Completion of reaction was verified by LC- MS. The solvent was removed and the resulting mixture was dissolved in methanol which was removed to give the second intermediate as a solid.
[00365] The mixture was dissolved in CH2Cl2 (150 mL) and washed with saturated
NaHCO3. The organic layer was dried over MgSO4, concentrated, and purified by silica gel chromatography three times, first using a methanol/CH2Cl2 gradient, the second time using a
NYI-4144519vl 86 hexane/ethyl acetate gradient followed by a methanol/ethyl acetate gradient, and a third time using a methanol/CH2Cl2 gradient.
[00366] C. To a suspension of the intermediate from Step 2B (0.1 10 g, 0.25 mmol) in
THF (5mL) was added Ph3P (0.079g, 0.3 mmol), diisopropylazodicarboxylate (0.06 Ig, 0.3 mmol) and 4-morpholinoethanol (0.039 g, 0.3 mmol). The reaction mixture was stirred at room temperature overnight. Completion of the reaction was verified by LC-MS. The solvent was removed and the final product was purified using silica gel chromatography, with methanol in CH2Cl2 (0.030g, 21%).
6.3 EXAMPLE 3. BULK SYNTHESIS OF N-(5-TERT-BUTYL- ISOXAZOL-3-YL)-N’-f4-[7-(2-MORPHOLIN-4-YL- ETHOXY^IMID AZO[2α-BUlJlBENZOTHIAZOL-2- YLlPHENYLiUREA (“COMPOUND Bl”)
[00367] A multi-step reaction scheme that was used to prepare bulk quantities of
Compound Bl is depicted in FIG. 66a and FIG. 66b, and is described further below. [00368] Step 1 : Preparation of 2- Amino-6-hydroxybenzothiazole (Intermediate 1). 2-
Amino-6-methoxybenzothiazole is reacted with hot aqueous HBr for about 3 hrs and then the clear solution is cooled to ambient temperature overnight. The precipitated solids are collected, dissolved in hot water and the pH is adjusted to between 4.5-5.5. The resultant solids are collected, dried and recrystallized from Isopropanol. Second crop material is collected. The solids are vacuum dried to give Intermediate 1.
[00369] Step 2: Preparation of 2-(4-Nitrophenyl) imidazo [2J-b]benzothiazol-7-ol
(Intermediate 2). 2-Amino-6-hydroxybenzothiazole, 2-Bromo-4-nitroacetophenone and absolute Ethanol are added together and heated to reflux for approximately 24 hours. Tetrabutylammonium iodide is added and the reaction is refluxed an additional 12 hours. The resulting yellow suspension is cooled and the solids collected and washed with Ethanol and Diethyl ether. The solids are dried under vacuum to give Intermediate 2. [00370] Step 3: Preparation of 7-(2-Morpholin-4-yl-ethoxy)-2-(4-nitrophenyl) imidazo
[2,1-b] benzothiazole (Intermediate 3). Intermediate 2, 4-(2-Chloroethyl)morpholine hydrochloride, Potassium carbonate and Tetrabutylammonium iodide are added to N,N- Dimethylformamide forming a yellow suspension that is heated for over 3 hours. The reaction is cooled and the solids are collected, slurried into water, filtered, slurried into
NYl-4 l4451′)v l 87 acetone, filtered and washed with Acetone to give yellow solids that are dried under vacuum to give Intermediate 3.
[O0371] Step 4: Preparation of 7-(2-Moφholin-4-yl-ethoxy)-2-(4-aminophenyl) imidazo [2,1 -b] benzothiazole (Intermediate 4). Intermediate 3 is dissolved into Methanol and THF and placed in a Hydrogenator. Raney Nickel is added and the vessel is pressurized with Hydrogen and stirred for >24 hrs. The reaction mixture is concentrated to a thick paste and diluted with Methyl tert-butyl ether. The resulting solids are filtered and washed with Methyl tert-butyl ether and dried under vacuum to give Intermediate 4. [O0372] Step 5: Preparation of {[5-(tert-Butyl) isoxazol-3-vnatnino}-N-{4-r7-(2- morpholin-4-yl-ethoxy)(4-hvdroimidazolo[2J-blbenzothiazol-2-yl)]phenyl|carboxamide (Compound Bl). 3 -Amino- 5 -tert-butyl isoxazole in Methylene chloride is added to a vessel containing toluene which is cooled to approx 0 0C. Triphosgene is then added and the reaction mixture is cooled to below -15 0C. Triethylamine is added, followed by Intermediate 4. The mixture is heated to distill off the Methylene chloride and then heated to over 60 0C for over 12 hours and cooled to 50-60 °C. The resulting solids are filtered, washed with Heptane, slurried with 4% sodium hydroxide solution, and filtered. The solids are then washed with Methyl tert-butyl ether followed by Acetone and dried under vacuum to give Compound Bl.
6.4 EXAMPLE 4. EXAMPLES OF PREPARATION OF COMPOUND Bl HCL SALT
[00373] Example A: For the preparation of a hydrochloride salt of Compound Bl5 N-
(5-tert-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,l- b][l,3]benzothiazol-2-yl]phenyl}urea hydrochloride, the free base was dissolved in a mixture of 20 ml methylene chloride and 1 ml methanol. A solution of 1.0 M HCl in ethyl ether (1.1 eq.) was added dropwise, followed by addition of ethyl ether. The precipitate was collected by filtration or centrirugation and washed with ethyl ether to give a hydrochloride salt of Compound Bl. Yield: 2.44 g (98 %) NMR (DMSO-^) S X 1.0 (br, IH), 9.68 (s, IH), 9.26 (s, IH), 8.66 (s, IH), 7.93 (d, IH), 7.78 (m, 3H), 7.53 (d, 2H), 7.26 (dd, IH), 6.53 (s, IH), 4.50 (t, 2H), 3.97 (m, 2H), 3.81 (t, 2H), 3.6 (overlapping, 4H), 3.23 (m, 2H), 1.30 (s, 9H). [00374] Example B: Concentrated HCl is added to a suspension of Compound Bl in warm methanol forming a solution that slowly begins to precipitate. The reaction mixture is
NYI-4144519vl 88 refluxed for over 2 hrs and then stirred overnight at ambient temperature. The HCl salt is collected and dried under vacuum.
[00375] Example C: Materials: {[5-(tert-Butyl) isoxazol-3-yl]amino}-N-{4-[7-(2- morpholin-4-yl-ethoxy)(4-hydroimidazolo[2,l-6]benzothiazol-2-yl)] phenyl }carboxamide (775 g, 1.38 mol, 1.0 eq); HCl 37% aqueous (288 mL, 3.46 mol, 2.5 eq); Methanol (MeOH, AR) (40L). Procedure: (Step 1) Equipped a 5OL 3-neck round bottom flask with a mechanical agitator, thermocouple probe, Nitrogen inlet, drying tube, reflux condenser, addition funnel and in a heating mantle. (Step 2) Charged the flask with {[5-(tert-Butyl) isoxazol-3-yl] amino}-N-{4-[7-(2-morpholin-4-yl-ethoxy)(4-hydroimidazolo[2,l- b]benzothiazol-2-yl)] phenyl jcarboxamide (775g) and MeOH, AR (40L). Heat the resulting off-white suspension to reflux (680C). A clear solution did not form. (Step 3) Added HCl (37% aqueous) (228 mL) over 5 minutes at 68°C. The reaction mixture turned into a clear solution and then a new precipitate formed within approximately 3 minutes. Continued heating at reflux for approximately 5 hours. Allowed the reaction mixture to cool to ambient temperature overnight. (Step 4) Collected the off-white solids by filtration onto a polypropylene filter, washing the solids with MeOH, AR (2 x 1 L). (Step 5) Combined two lots of material prepared in this manner (74Og and 82Og). Slurried the combined solids in Methanol (30 L) over 30 minutes at reflux and cool to the room temperature. (Step 6) Collected the solids by filtration onto a polypropylene filter, rinsing with Methanol (2 x 1.5L). (Step 7) Dried the solids in a vacuum oven (<10mniHg) at 400C. Yield: 1598 g (84%), off-white solid; HPLC: 98.2% (area); MS: 561.2 (M+l); IH NMR: conforms (300 MHz, DMSO-d6); Elemental Analysis (EA): Theory = 54.97 %C; 5.41 %H; 13.26 %N; 5.06 %S; 11.19 %C1; Actual = 54.45 %C; 5.46 %H; 13.09 %N; 4.99 %S; 10.91 %C1.
NYl-4I44519v! 89 [00376] Examples of Compound Bl HCl salt synthesis

[00377] Example D: In a 50-L 3-neck round bottom flask equipped with a mechanical stirrer, heating mantle, condenser and nitrogen inlet was charged Compound Bl (1052.4 g, 1.877 mol, 1.00 equiv.) and methanol (21 L). The reactor was heated and stirred. At an internal temperature > 50 0C, cone. HCl (398.63 mL, 4.693 mol, 2.5 equiv.) was charged over 5 minutes through an addition funnel. With the addition, the reaction changed from a pale yellow suspension to a white suspension. The internal temperature was 55 0C at the conclusion of the addition. The reaction was heated to reflux for 1 hour, then heating discontinued and the reaction allowed to cool to room temperature. The reaction was filtered in two portions, each filter cake washed with methanol (2 x 1 L), transferred to trays and dried in a vacuum oven (45 0C) to constant weight. The dried trays were combined to produce 1141.9 g, 96% yield, 99.1 % HPLC purity, 10.9% chloride by titration.
Solid Forms Comprising the HCl Salt of Compound Bl 6.6.2.1 Preparation of Solid Forms

6.6.2.2 Cold Precipitation Experiments

NYl-4144519vl 102 6.6.2.3 Slurry Experiments
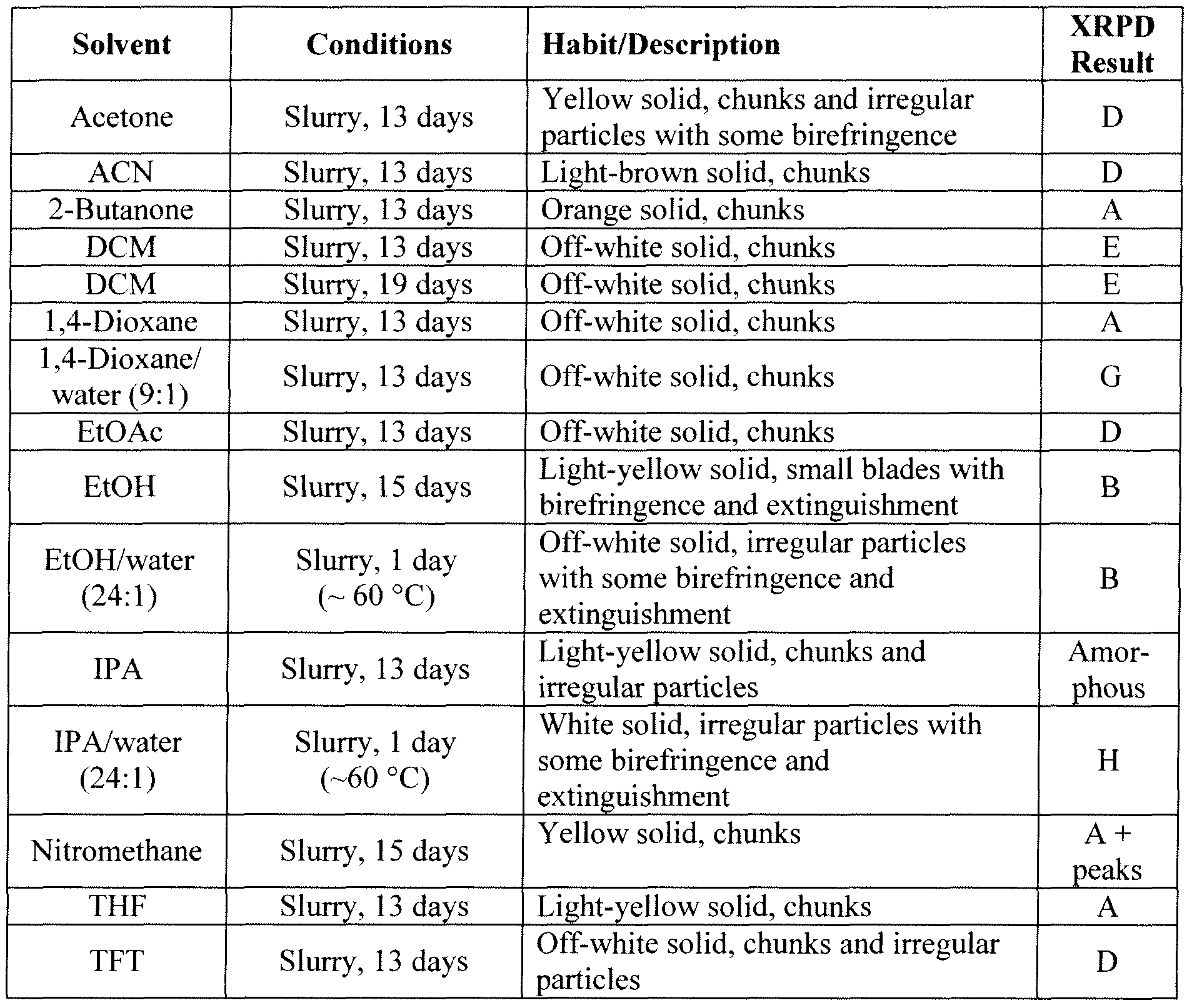
NYI-41445 l9vl 103 6.6.2.4 Additional Preparation of Solid Forms Comprising the HCI Salt of Compound Bl

NYl-4144519v l 104

NYM 144519vl 105

N Y l -4 1 4 4 5 1 9 v l 1 0 6

NYI-4I44519vi 107

N V I 4 1 4 4 5 1 9 1 0 8
“Abbreviations in Table: CC = crash cool, CP = crash precipitation, EtOAc = ethyl acetate, FE = fast evaporation, VD = vapor diffusion, IPA = isopropanol, MEK = methyl ethyl ketone (2-butanone), RE = rotary evaporation, RT = room (ambient) temperature, SC = slow cool, SE = slow evaporation, THF = tetrahydrofuran, TFE = 2,2,2=trifluoroethanol.
6.6.2.5 Scale-up Experiments of Involving Crystal Forms Comprising the HCl Salt of Compound Bl

NYI-4144519v l 109

Abbreviations in Table: CC = crash cool, CP = crash precipitation, EtOAc = ethyl acetate, FE = fast evaporation, IPA = isopropanol, MEK = methyl ethyl ketone (2-butanone), RE = rotary evaporation, RT = room (ambient) temperature, SC = slow cool, SE = slow evaporation, THF = tetrahydrofuran, TFE = 2,2,2=trifluoroethanol.
……………………
Identification of N-(5-tert-butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,1-b][1,3]benzothiazol-2-yl]phenyl}urea dihydrochloride (AC220), a uniquely potent, selective, and efficacious FMS-like tyrosine kinase-3 (FLT3) inhibitor
J Med Chem 2009, 52(23): 7808
J Med Chem 2009, 52(23): 7808
N-(5-tert-Butyl-isoxazol-3-yl)-N′-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,1-b][1,3]benzothiazol-2-yl]phenyl}urea Dihydrochloride (7): General Procedure D
A suspension of 2-(4-aminophenyl)-7-(2-morpholin-4-yl-ethoxy)imidazo[2,1-b][1,3]benzothiazole (19c) (4.06 g, 10.3 mmol) and 5-tert-butyl-isoxazole-3-isocyanate (5) (1.994 g, 12 mmol) in toluene (60 mL) was heated at 120 °C overnight. The reaction was quenched with a mixture of dichloromethane and water containing a little methanol, and the mixture was neutralized with saturated aqueous NaHCO3. The aqueous phase was extracted twice with dichloromethane, and the combined organic extracts were dried over MgSO4 and filtered. The filtrate was concentrated to a volume of about 20 mL and ethyl ether was added, resulting in the formation of a solid. The precipitate was collected by filtration, washed with ethyl ether, and dried under vacuum to give the free base of 7 (2.342 g, 41%).
1H NMR (DMSO-d6) δ 9.6 (br, 1H), 8.9 (br, 1H), 8.61 (s, 1H), 7.86 (d, J = 8.9 Hz, 1H), 7.76 (d, J = 8.0 Hz, 2H), 7.69 (d, J = 1.3 Hz, 1H), 7.51 (d, J = 8.0 Hz, 2H), 7.18 (dd, J = 1.3 and 8.9 Hz, 1H), 6.52 (s, 1H), 4.16 (t, J = 5.7 Hz, 2H), 3.59 (t, J = 4.2 Hz, 4H), 3.36 (overlapping, 4H), 2.72 (t, J = 5.7 Hz, 2H), 1.30 (s, 9H).
General Procedure E for Preparation of Hydrochloride Salt
The free base was dissolved in a mixture of dichloromethane (20 mL) and methanol (1 mL). A solution of 1.0 M HCl in ethyl ether (1.1 equiv for all compounds except 7, for which 2.5 equiv were used) was added dropwise, followed by addition of ethyl ether. The precipitate was collected by filtration to give
N-(5-tert-butyl-isoxazol-3-yl)-N′-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,1-b][1,3]benzothiazol-2-yl]phenyl}urea dihydrochloride (7) (2.441 g, 98%).
1H NMR (DMSO-d6) δ 11.0 (br, 1H), 9.68 (s, 1H), 9.26 (s, 1H), 8.66 (s, 1H), 7.93 (d, J = 8.9 Hz, 1H), 7.78 (m, 3H), 7.53 (d, J = 8.7 Hz, 2H), 7.26 (dd, J = 2.4 and 8.9 Hz, 1H), 6.53 (s, 1H), 4.50 (t, J = 4.1 Hz, 2H), 3.97 (m, 2H), 3.81 (t, J = 12.1 Hz, 2H), 3.6 (overlapping, 4H), 3.23 (m, 2H), 1.30 (s, 9H). LC-MS (ESI) m/z 561 (M + H)+.
Anal. (C29H32N6O4S·2HCl) C, H, N. C: calcd 54.97; found 54.54. H: calcd 5.22; found 5.87. N: calcd 13.26; found 13.16.
References
- Chao, Qi; Sprankle, Kelly G.; Grotzfeld, Robert M.; Lai, Andiliy G.; Carter, Todd A.; Velasco, Anne Marie; Gunawardane, Ruwanthi N.; Cramer, Merryl D.; Gardner, Michael F.; James, Joyce; Zarrinkar, Patrick P.; Patel, Hitesh K.; Bhagwat, Shripad S. (2009). “Identification of N-(5-tert-Butyl-isoxazol-3-yl)-N’-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,1-b][1,3]benzothiazol-2-yl]phenyl}urea Dihydrochloride (AC220), a Uniquely Potent, Selective, and Efficacious FMS-Like Tyrosine Kinase-3 (FLT3) Inhibitor”. Journal of Medicinal Chemistry 52 (23): 7808–7816.
- Drug Tames Refractory AML. ASH Dec 2012
- NMR……….http://file.selleckchem.com/downloads/nmr/S152601-AC-220-HNMR-Selleck.pdf
- HPLC………http://file.selleckchem.com/downloads/hplc/S152601-AC-220-HPLC-Selleck.pdf
18 BOSUTINIB
BOSUTINIB
4-[(2,4-dichloro-5-methoxyphenyl)amino]-6-methoxy-7-[3-(4-methylpiperazin-1-yl)propoxy]quinoline-3-carbonitrile
Bosutinib Monohydrate (伯舒替尼)
(Bosulif®)
Approved sept4 2012 by FDA
PMDA SEPT26 2014
EMA MAR 27 2013
A kinase inhibitor indicated for the treatment of adult patients with Ph+ chronic myelogenous leukemia (CML).WYETH INNOVATOR
PFIZER DEVELOPER
SKI-606; SK-606
CAS No.380843-75-4 (Free form)
CAS 918639-08-4(Bosutinib Monohydrate)
Bosutinib (rINN/USAN; codenamed SKI-606, marketed under the trade name Bosulif) is atyrosine kinase inhibitor undergoing research for use in the treatment of cancer. [1] [2]Originally synthesized by Wyeth, it is being developed by Pfizer.
。


Some commercial stocks of bosutinib (from sources other than the Pfizer material used for clinical trials) have recently been found to have the incorrect chemical structure, calling the biological results obtained with them into doubt.[3]
Bosutinib received US FDA approval on September 5, 2012 for the treatment of adult patients with chronic, accelerated, or blast phase Philadelphia chromosome-positive (Ph+)chronic myelogenous leukemia (CML) with resistance, or intolerance to prior therapy.[4][5][6]
Article
Good News For Pfizer’s Orphan Drug Bosulif (Bosutinib) in Europe
January 18, 2013
The European Medicines Agency’s (EMA) Committee for Medicinal Products for Human Use (CHMP) on January 17, 2013, adopts a positive opinion, recommending a conditional marketing authhorization for Pfizer’s orphan drug Bosulif (Bosutinib) for Chronic Leukemia (CML). Bosutinib receives orphan designation from the European Commission (EC) on August 4, 2010, for CML.
Pfizer receives FDA approval on September 4, 2012, for orphan drug Bosulif (Bosutinib) for CML. Pfizer receives on February 24, 2009, FDA Orphan Drug Designation (ODD) for Bosutinib for CML.
Per a September 2012 article in FierceBioTech.com, a Pfizer spokesperson says that “the drug will cost less than $8,200/month”/patient in the US. In other words, treatment will cost approximately $98,400/patient/year. Also per FierceBiotech,“Bosulif is the 3rd new medicine from Pfizer Oncology’s pipeline to be approved by the FDA in just 13 months ….”.
ARTICLE
Pfizer’s response to compound fraud spotlights quality issues
read .........http://www.rsc.org/chemistryworld/2015/12/pfizer-bogus-bosutinib-isomer-fraud-leukaemia-drug
Synthesis












REFERENCES
- Puttini M, Coluccia AM, Boschelli F, Cleris L, Marchesi E, Donella-Deana A, Ahmed S, Redaelli S, Piazza R, Magistroni V, Andreoni F, Scapozza L, Formelli F, Gambacorti-Passerini C. In vitro and in vivo activity of SKI-606, a novel Src-Abl inhibitor, against imatinib-resistant Bcr-Abl+ neoplastic cells. Cancer Res. 2006 Dec 1;66(23):11314-22. Epub 2006 Nov 17.
- Vultur A, Buettner R, Kowolik C, et al. (May 2008). "SKI-606 (bosutinib), a novel Src kinase inhibitor, suppresses migration and invasion of human breast cancer cells".Mol. Cancer Ther. 7 (5): 1185–94. doi:10.1158/1535-7163.MCT-08-0126.PMC 2794837. PMID 18483306.
- Derek Lowe, In The Pipeline (blog), "Bosutinib: Don't Believe the Label!"
- Cortes JE, Kantarjian HM, Brümmendorf TH, Kim DW, Turkina AG, Shen ZX, Pasquini R, Khoury HJ, Arkin S, Volkert A, Besson N, Abbas R, Wang J, Leip E, Gambacorti-Passerini C. Safety and efficacy of bosutinib (SKI-606) in chronic phase Philadelphia chromosome-positive chronic myeloid leukemia patients with resistance or intolerance to imatinib. Blood. 2011 Oct 27;118(17):4567-76. Epub 2011 Aug 24.
- Cortes JE, Kim DW, Kantarjian HM, Brümmendorf TH, Dyagil I, Griskevicus L, Malhotra H, Powell C, Gogat K, Countouriotis AM, Gambacorti-Passerini C. Bosutinib Versus Imatinib in Newly Diagnosed Chronic-Phase Chronic Myeloid Leukemia: Results From the BELA Trial. J Clin Oncol. 2012 Sep 4. [Epub ahead of print]
- "Bosulif Approved for Previously Treated Philadelphia Chromosome-Positive Chronic Myelogenous Leukemia". 05 Sep 2012.
- P Bowles et al, Org. Process Res. Dev., 2015, DOI: 10.1021/acs.oprd.5b00244
8 N M Levinson and S G Boxer, PLoS One, 2012, 7, e29828 (DOI: 10.1371/journal.pone.0029828)
9 N Beeharry et al, Cell Cycle, 2014, 13, 2172 (DOI: 10.4161/cc.29214)
19
Nintedanib esylate is a potent triple angiokinase inhibitor for VEGFR1/2/3, FGFR1/2/3 and PDGFRα/β. BIBF 1120 esylate
ReplyDelete