
SILODOSIN
Urief, 160970-54-7, Rapaflo, KMD 3213, Silodyx, KAD 3213, KMD-3213
Molecular Formula: C25H32F3N3O4
Molecular Weight: 495.53449 g/mol
Alpha 1A adrenoceptor antagonist
Prostate hyperplasia
Kissei Pharmaceutical Co Ltd INOVATOR
CAS 160970-54-7
2,3-Dihydro-1-(3-hydroxypropyl)-5-[(2R)-2-[[2-[2-(2,2,2-trifluoroethoxy)phenoxy]ethyl]amino]propyl]-1H-indole-7-carboxamide
160970-64-9 (racemate)
169107-04-4 (diHBr)
169107-04-4 (diHBr)
Properties: [a]D25 -14.0° (c = 1.01 in methanol).
Optical Rotation: [a]D25 -14.0° (c = 1.01 in methanol)
Therap-Cat: In treatment of benign prostatic hypertophy.
a-Adrenergic Blocker.
In February 2008, the FDA accepted for review an NDA for silodosin for the treatment of dysuria associated with BPH . In October 2008, the FDA approved the drug . In April 2009, Actavis launched silodosin for the treatment of the signs and symptoms of BPH .
 SILODOSIN
SILODOSIN
1-(3-hydroxypropyl)-5-[(2R)-2-[2-[2-(2,2,2-trifluoroethoxy)phenoxy]ethylamino]propyl]-2,3-dihydroindole-7-carboxamide
Patent (5387603 United States)
Patent (5403847 United States)
Kissei Pharmaceutical, Daiichi Sankyo (formerly Daiichi Seiyaku), Actavis (formerly Watson) and Recordati have developed and launched silodosin (Urief; Trupass; Rapaflo; Thrupas; Silodyx; Urorec; KMD-3213; Youlifu), an oral alpha 1A adrenoceptor antagonist selective for prostatic receptors . The product is comarketed in Europe by several licensees. The drug is indicated for the treatment of the signs and symptoms of benign prostatic hyperplasia (BPH).
Silodosin, a highly selective alpha1A-adrenoceptor antagonist, was launched in May 2006 in Japan for the oral treatment of urinary disturbance associated with benign prostatic hyperplasia (BPH). The product was launched in the U.S. for the treatment of signs and symptoms of benign prostatic hyperplasia in 2009. In 2009, a positive opinion was received in the E.U. for this indication and final approval was obtained in 2010. Launch in the E.U. took place the same year.
In May 2006, silodosin was launched as a capsule formulation in Japan. Actavis launched the drug in the US in April 2009. In June 2010, EU launched began, initially with Germany ; in November 2010, the drug was launched in France; by December 2010, the drug was launched in Spain.
In 2001, Kissei established an agreement with Daiichi Pharmaceutical to codevelop and comarket silodosin. An oral, once-daily formulation of silodosin filed in the U.S. by Watson (now Actavis) was approved in 2008. Watson (now Actavis) obtained exclusive rights in 2004 to develop and market the drug in the U.S.
PRODUCT Was developed and launched byKissei Pharmaceutical, Daiichi Sankyo, Actavis and Recordati. Family members of the product case EP0600675 have SPC protection in most EU states until 2018; while its Orange Book listed equivalent, US5387603, expire in the US in 2018 with US156 extension.
Silodosin (trade names Rapaflo (USA), Silodyx (Europe and South Africa), Rapilif (India), Silodal (India), Urief (Japan), Urorec (Russia)) is a medication for the symptomatic treatment of benign prostatic hyperplasia. It acts as an α1-adrenoceptor antagonist with high uroselectivity (selectivity for the prostate).
 | |
| Systematic (IUPAC) name | |
|---|---|
| 1-(3-hydroxypropyl)-5-[(2R)-({2-[2-[2-(2,2,2-trifluoroethoxy)phenoxy]ethyl}amino)propyl]indoline-7-carboxamide | |
| Clinical data | |
| |
| Legal status |
|
| Routes | Oral |
| Pharmacokinetic data | |
| Bioavailability | 32% |
| Protein binding | 97% |
| Metabolism | Hepatic glucuronidation (UGT2B7-mediated); also minor CYP3A4 involvement |
| Half-life | 13±8 hours |
| Excretion | Renal and fecal |
| Identifiers | |
| CAS number | 160970-54-7 |
| ATC code | G04CA04 |
| PubChem | CID 5312125 |
| IUPHAR ligand | 493 |
| ChemSpider | 4471557 |
| UNII | CUZ39LUY82 |
| ChEMBL | CHEMBL24778 |
| Synonyms | KAD-3213, KMD-3213 |
| Chemical data | |
| Formula | C25H32F3N3O4 |
| Molecular mass | 495.534 g/mol |
History
Silodosin received its first marketing approval in Japan in May 2006 under the tradename Urief, which is jointly marketed by Kissei Pharmaceutical Co., Ltd. and Daiichi Sankyo Pharmaceutical Co., Ltd.
Kissei licensed the US, Canadian, and Mexican rights for silodosin to Watson Pharmaceuticals, Inc. in 2004.
On February 12, 2008, Watson announced that the New Drug Application submitted to the United States Food and Drug Administration for silodosin has been accepted for filing. FDA approved this drug on October 9, 2008.[1] Silodosin is marketed under the trade names Rapaflo in the US and Silodyx in Europe.[2] and Rapilif in India (Ipca Urosciences)
Pharmacology
Since silodosin has high affinity for the α1A adrenergic receptor, it causes practically no orthostatic hypotension (in contrast to other α1 blockers). On the other side, the high selectivity seems to cause more problems with ejaculation.[3]
As α1A adrenoceptor antagonists are being investigated as a means to male birth control due to their ability to inhibit ejaculation but not orgasm, a trial with 15 male volunteers was conducted. While silodosin was completely efficacious in preventing the release of semen in all subjects, 12 out of the 15 patients reported mild discomfort upon orgasm. The men also reported the psychosexual side effect of being strongly dissatisfied by their lack of ejaculation.[4]

///////////////////////////////
CN 103848772
silodosin (Silodosin) is 〃 2 Japanese orange Johnson invented - receptor antagonist, for the treatment of benign prostatic hyperplasia or hypertrophy, and other related symptoms. Clinical trials showed that 25% of patients with benign prostatic hyperplasia need for drugs or surgery. Although prostatectomy is better, the mortality rate is not high, but patients bring varying degrees of damage. So look for an effective and safe non-surgical treatment, not only can control the further development of the disease, while relieving the symptoms of the patient.
benign prostatic hyperplasia in older male patients have a higher prevalence, and clinical alternative drugs rarely, so the development of a benign prostatic hyperplasia drug treatment, not only has good social benefits, but also to bring good economic benefits. The study confirmed that silodosin is the treatment of benign prostatic hyperplasia in an important class of drugs.

Currently, the research reported in the published literature on the preparation of compounds of silodosin, are:


Early 1995, Kitazawa M et al patent US5387603, the reporter silodosin total synthesis method, but the method reaction step is long, the yield is not too high, not suitable for our industrial production.

In 2009, 翟富民 et al patent CN102115455A, which reported a method for preparing Sailuoduoxin key intermediates. The appropriate method for improving existing methods, although shorter than the previous method step in the step, but low synthesis yield of the process, we can not meet the needs of industrial production.
In summary, the compounds prepared silodosin more synthetic methods are constantly improved, but there are still a lot of flaws. Therefore, there is need for further research on the preparation of compounds of silodosin to get simple process, product yield, product easy separation of the new preparation.SUMMARY
The present invention is to overcome the above problems of the prior art, there is provided a method for preparing important intermediates silodosin, the present invention is simple process, high yield, easy separation of the product, the method suitable for industrial production .
To achieve the above technical object, to achieve the above technical result, the present invention is realized by the following technical scheme:
One kind of silodosin preparation of important intermediate, comprising the steps of:
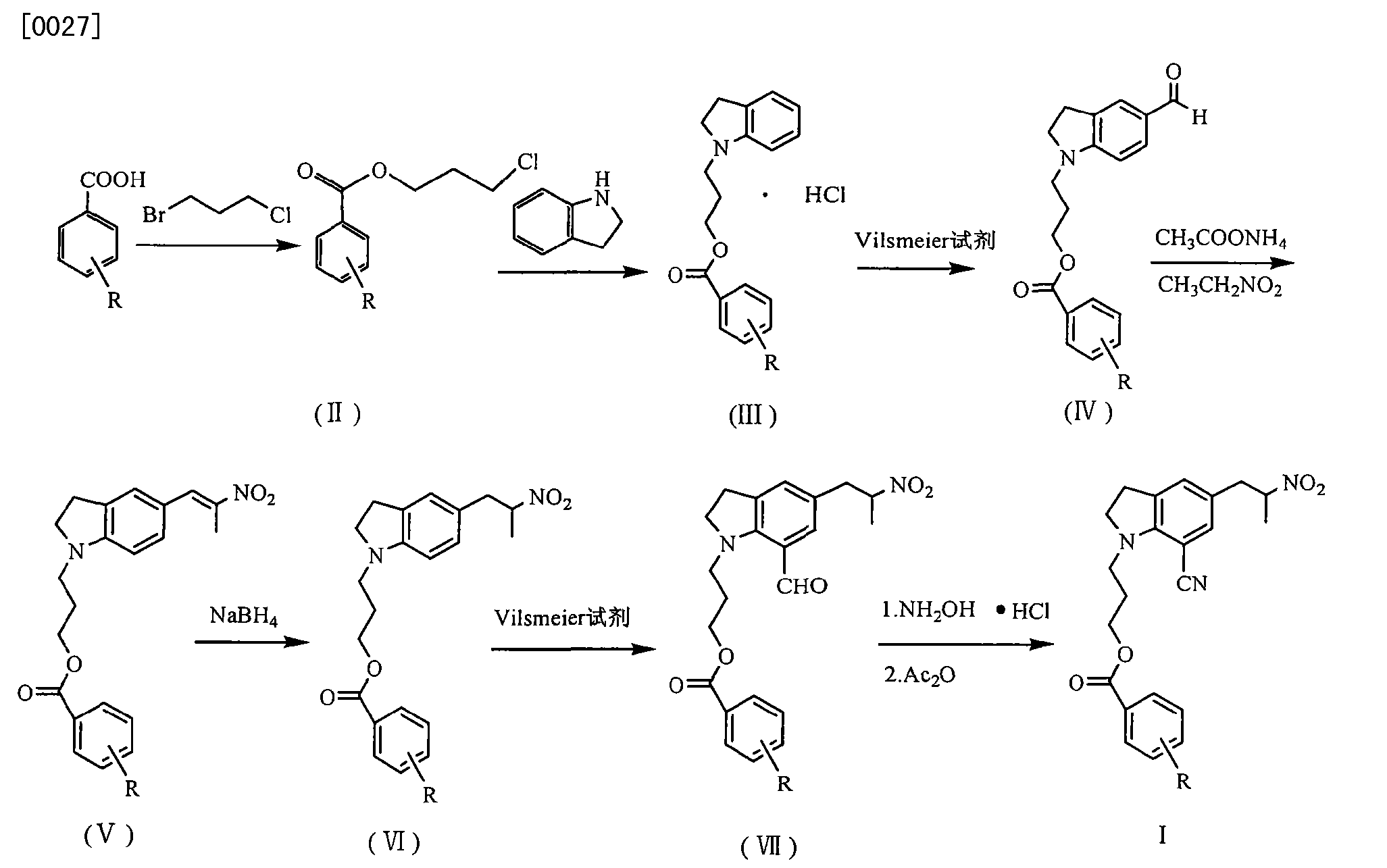
Step I) in a flask, 282g of raw materials 1-acetyl-5- (2-bromo-propyl) indoline, 222g phthalimide potassium salt and 700mL DMF, was heated at 110 ° C for 2h; After completion of the reaction, to which was added the right amount of water to wash away the excess solvent DMF and salt extraction desolventizing after EA, was 296g crude;
Step 2) In a flask was added 296g crude product obtained in step I, dissolved 800mL ethanol, was added 165mL of hydrazine hydrate, 50 ° C is heated to precipitate a white solid; After completion of the reaction, cooling suction filtered, the filter cake washed with ethanol, and then the mother liquor removing solvent under reduced pressure; After dissolving EA, washed with water to wash away the excess hydrazine, and finally the organic phase the solvent was removed to give 165g intermediate, i.e. 1-acetyl-5- (2-aminopropyl) indoline;
Step 3) In the three-necked flask, 165g of Intermediate 1-acetyl-5- (2-aminopropyl) indoline, dissolved 600mL methanol, stirred at room temperature, and thereto was slowly added dropwise bromine; the addition was complete After stirring at room temperature 5-6h; After completion of the reaction, slowly poured into saturated NaHSO3, and wash away excess bromine; extracted with ethyl acetate, washed with water and saturated brine, dried over anhydrous sodium sulfate; After filtration, the solvent was removed in vacuo to give the crude product recrystallized from toluene to give 177g pure product;
Step 4) In a flask was added 177g of pure product obtained in Step 3 and 65g CuCN, after use 700mL DMF, was heated at 110 ° C for 3 to 5h; After completion of the reaction, the amount of water was added thereto, washing off excess solvent DMF and salt, EA desolventizing crude extract, after recrystallization 121g pure 1-acetyl-5- (2-bromo-propyl) -7-cyano-indoline that silodosin important intermediates;
Step (1), (2), (3), (4) synthesis reaction is:

Further, the step I) to step 4) by TLC plate tracking point detection reaction.
The beneficial effects of the present invention are:
Preparation silodosin important intermediate of the present invention, mention of the method is simple, high reaction yield, product easily separated, suitable for industrial production and so on.
Preparation Method A silodosin important intermediate, comprising the following steps: Step I) in a flask, 282g of raw materials 1-acetyl-5- (2-bromo-propyl) indoline, 222g o phthalimide potassium and 700mL DMF, heated at 110 ° C for 2h; After completion of the reaction, to which was added the right amount of water to wash away the excess solvent DMF and salt extraction desolventizing after EA, was 296g crude;
Step 2) In a flask was added 296g crude product obtained in step I, dissolved 800mL ethanol, was added 165mL of hydrazine hydrate, 50 ° C is heated to precipitate a white solid; After completion of the reaction, cooling suction filtered, the filter cake washed with ethanol, and then the mother liquor removing solvent under reduced pressure; After dissolving EA, washed with water to wash away the excess hydrazine, and finally the organic phase the solvent was removed to give 165g intermediate, i.e. 1-acetyl-5- (2-aminopropyl) indoline;
Step 3) In the three-necked flask, 165g of Intermediate 1-acetyl-5- (2-aminopropyl) indoline, dissolved 600mL methanol, stirred at room temperature, and thereto was slowly added dropwise bromine; the addition was complete After stirring at room temperature 5-6h; After completion of the reaction, slowly poured into saturated NaHSO3, and wash away excess bromine; extracted with ethyl acetate, washed with water and saturated brine, dried over anhydrous sodium sulfate; After filtration, the solvent was removed in vacuo to give the crude product recrystallized from toluene to give 177g pure product;
Step 4) In a flask was added 177g of pure product obtained in Step 3 and 65g CuCN, after use 700mL DMF, was heated at 110 ° C for 3 to 5h; After completion of the reaction, the amount of water was added thereto, washing off excess solvent DMF and salt, EA desolventizing crude extract, after recrystallization 121g pure 1-acetyl-5- (2-bromo-propyl) -7-cyano-indoline that silodosin important intermediates;
Step (1), (2), (3), (4) synthesis reaction is:

Further, the step I) to step 4) by TLC plate tracking point detection reaction.
.........................................................
WO2013056842
Silodosin is commercially available under the tradenames RAPAFLO® or
UROPvEC as a capsule formulation for oral use containing 4 mg or 8 mg of the drug. The capsules are to be taken orally once daily for the treatment of the signs and symptoms of benign prostatic hyperplasia. US 5,387,603 and EP 0 600 675 disclose silodosin as a therapeutic agent for the treatment for dysurea associated with benign prostatic hyperplasia. The molecular structure of silodosin (XXV) is shown below.
(XXV)

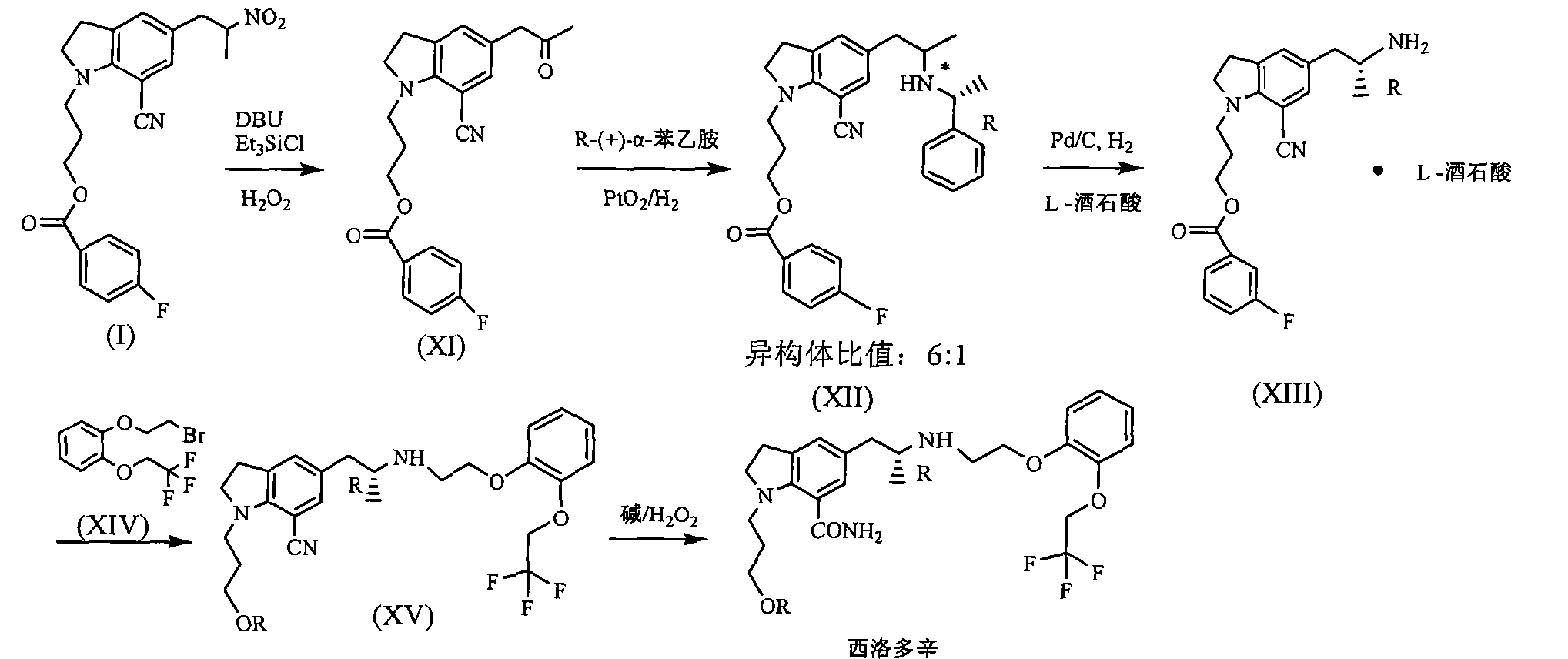
The synthesis of silodosin is relatively complex and requires a sequence of multiple steps. A key intermediate compound in the synthesis of silodosin is the optically active amine compound represented by the general formula R-Y:

1
wherein, R represents a protecting group and R represents a cyano (CN) or carbamoyl (CONH2) group. The intermediate compound R-Y bears the asymmetric carbon atom that imparts the optical activity to silodosin. Therefore, it is important to obtain the compound R-Y with high optical purity, because according to the methods reported in the state of the art the optical purity of the compound R-Y determines the optical purity of the final product silodosin.
JP 2001-199956 discloses a process for the preparation of a compound of formula R-Y, wherein l-(3-benzoyloxypropyl)-7-cyano-5-(2-oxopropyl)-2,3- dihydroindole or the corresponding 7-carbamoyl derivative is reacted with an optically active amine, namely L-2-phenylglycinol or L-l-phenylethanamine, to afford an imine compound of formula III as depicted in the below scheme 1. Scheme l . JP 2001-199956

R1 = COPh; R2 = CN or CONH2; R3 = H or OH a = 1. cat. deprotection
2. frational crystallization with L-tartaric acid
b = 1. chromatographic separation
2. cat. deprotection
The optically active imine III is subjected to catalytic hydrogenation using platinum(IV) oxide as a catalyst affording the diastereomers IV in a ratio of 3.8:1. The chiral auxiliary II is subsequently removed by catalytic hydrogenation using 10% palladium on carbon, i. e. under the typical conditions which lead to the cleavage and removal of benzylic protecting groups from nitrogen or oxygen atoms. The catalytic deprotection reaction affords the desired intermediate compound R-Y with an optical purity corresponding to the ratio of the diasteromers obtained in the previous step, i. e. the ratio of compound R-Y to S-Y is approximately 3.8: 1, which corresponds to an optical purity of approximately 58.3% enantiomeric excess (e.e.).
In order to increase the optical purity of the intermediate R-Y JP 2001-199956 suggests to conduct a fractional crystallization of the desired enantiomer with L-tartaric acid. After a series of fractional crystallizations the compound R-Y is obtained with an optical purity of 97.6% enantiomeric excess. Alternatively, the diastereomers of the compound of formula IV are separated using chromatographic techniques as column chromatography on silicagel. The pure diastereomer R-TV affords the desired enantiomer R-Y with an optical purity of 100% e.e. after removal of the chiral auxiliary II with hydrogen using 10% palladium on carbon as catalyst.
Another approach for the synthesis of the key intermediate compound R-Y is reported in JP 2002-265444. The route of synthesis disclosed in said document is depicted in the below scheme 2.
Scheme 2. JP 2002-265444


R1 = CH2Ph (Bn); R2 = CN The process involves the reaction of an enantiomeric mixture of the compound of formula VI with (I S, 2R)-2-benzylaminocyclohexane methanol (VII) to obtain a diastereomeric mixture containing the salt VIII. After a series of crystallizations the diastereomer VIII was obtained with an optical purity of 92.8% diastereomeric excess (d.e.). Subsequently, the salt VIII was treated with an acidic aqueous solution to release the acid R-Vl from the salt. After extraction from the aqueous solution with ethyl acetate the acid R-Vl is converted into its amide IX. The compound IX is finally subjected to a Hofmann type rearrangement reaction to obtain the desired intermediate compound R-V.
WO 201 1/030356 discloses a process for the preparation of the intermediate compound R-V, which avoids the resolution of the enantiomers of specific intermediate compounds using chiral auxiliaries or optically active bases. The route of synthesis described in WO 201 1/030356 starts from L-alanine (X), which is a naturally occurring optically active amino acid. The process described in
WO 2011/030356 is depicted in the below scheme 3.

R1 = trimethylsilyl (TMS), tert-butyl dimethylsilyl (TBDMS), allyl, benzyl, propargyl R2 = CN or CONH2 The amino acid is protected by the addition of ethyl chloroformate and subsequently activated by the addition of oxalyl chloride to afford i?-(N-ethoxycarbonyl)alanine as an acyl chloride (XI). Said acyl chloride is reacted with hydroxy protected l-(3- hydroxypropyl)-7-cyano-2,3-dihydroindole of formula XII in a Friedel-Crafts acylation reaction, which gives a compound of formula XIII. The oxo group in compound XIII is reduced to afford a compound of formula XIV that is subsequently subjected to a hydrolysis reaction to yield the key intermediate compound R-Y. It is an object of the present invention to provide a process for preparing silodosin or a pharmaceutically acceptable salt thereof, which process affords the drug with high optical purity and with better yield compared to the prior art processes. This object is solved by the subject matter as defined in the claims.
Scheme 5. Conversion of com ound V to silodosin

R = protecting group
R2 = CN or CONH2
X = leaving group
Example 11. Silodosin (XXV)
A. The compound XXIV (18.0 g) was dissolved in methanol (150 ml) and 5% aqueous sodium hydroxide solution (50 ml). The reaction mixture was stirred at room temperature for 2 h. The deprotected compound XXIV, i. e. a compound of formula XXIV with R = hydrogen and R = cyano, was extracted with toluene. Subsequently, a 10% lactic acid solution (25 ml) was added to the toluene phase in order to extract the product in the aqueous phase. The aqueous solution was separated and then basified. The deprotected product was finally extracted with ethyl acetate. Removal of the solvent gives the deprotected compound to XXIV (R1 = H and R2 = CN; 1 1.0 g) as an oily mass.
B. A mixture of compound XXIV (R1 = H and R2 - CN; 10.0 g), DMSO (80 ml) and 5N NaOH solution (9.0 ml) was stirred for 15 min. at room temperature. An aqueous H202 (30%) solution (1 1.0 ml) was added to the reaction mixture, which was stirred at room temperature for additional 2 h after completion of the addition. Water was added to the reaction mixture, the product was extracted with ethyl acetate, and the solvent was subsequently evaporated to afford 9.0 g crude silodosin.
Example 12. Silodosin (XXV)
10.0 g of crude silodosin (optical purity = 85.0% e.e.) was dissolved in ethyl acetate (120 ml) at 55°C. The resulting clear solution was gradually cooled to 25°C under stirring. The suspension was further cooled to 15°C and stirred for 2 hours. The precipitated solid was filtered and dried at 50°C under vacuum to obtain 7.2 g of XXV with an optical purity of 97.5% e.e.
Example 13. Silodosin (XXV)
10.0 g of crude silodosin (optical purity = 98.5% e.e.) was dissolved in ethyl acetate (120 ml) at 55°C. The resulting clear solution was gradually cooled to 25 °C under stirring. The suspension was further cooled to 15°C and stirred for 2 hours. The precipitated solid was filtered and dried at 50°C under vacuum to obtain 7.2 g of XXV with an optical purity of 99.9% e.e.
Example 14. Silodosin (XXV)
10.0 g of crude silodosin (optical purity = 90.0 %e.e.) was dissolved in ethyl acetate (120 ml) at 55°C. The resulting clear solution was gradually cooled to 25°C under stirring. The suspension was further cooled to 15°C and stirred for 2 hours. The precipitated solid was filtered and dried at 50°C under vacuum to obtain 7.2 g of XXV with an optical purity of 97.0% e.e.
Example 15. Silodosin (XXV)
10.0 g of crude silodosin (optical purity = 92.0% e.e.) was dissolved in isopropyl acetate (160 ml) at 55°C. The resulting clear solution was gradually cooled to 25°C under stirring. The suspension was further cooled to 15°C and stirred for 2 hours. The precipitated solid was filtered and dried at 50°C under vacuum to obtain 8.2 g of XXV with an optical purity of 98.0% e.e. Example 16. Silodosin (XXV)
10.0 g of crude silodosin (optical purity = 98.0% e.e.) was dissolved in isopropyl acetate (160 ml) at 55°C. The resulting clear solution was gradually cooled to 25°C under stirring. The suspension was further cooled to 15°C and stirred for 2 hours. The precipitated solid was filtered and dried at 50°C under vacuum to obtain 8.0 g of XXV with an optical purity of 99.5% e.e.
....................................
EP2475634
Scheme- 1.

Scheme-2.

Scheme-3.

Scheme-4.

Scheme-5.

Example-14
Preparation of Preparation of l-(3-Hydroxy-propyl)-5-(2(R)-{2-[2-(2, 2, 2-trifluoro- ethoxy)-phenoxy]-ethyIamino}-propyl)-2,3-dihydro-lH-indol-7-carboxylic acid amide (I)(Silodosin)
To a solution of Benzoic acid 3-[5(R)-(2-amino-propyl)-7-cyano-2, 3-dihydro-indol-l- yl]-propyl ester (XV) (3.5 g, 10 mmole) in Dimethyl sulphoxide (60 ml), charged Hydrogen peroxide (10% w/w) (11 ml). Then added 5 N sodium hydroxide solution (12.3 ml) and reaction mass was stirred for 2 hours. After completion of reaction water was added and extracted the product in ethyl acetate. Organic layer was washed with brine and dried over sodium sulphate. The solvent was evaporated below 40°C under reduced pressure and added methanol (25 ml). To this solution charged glacial acetic acid (0.25 g, 4mmole) and [2-(2, 2, 2-Trifluoro-ethoxy)-phenoxy]-acetaldehyde (VIII) (3 g, 0.0125 mole). Reaction mixture was stirred at 25-30°C for 1 hour. Then reacted with sodium cyanoborohydride (0.15 g, 2.8 mmoles) and heated at 40-45°C for 2 hours. After the completion of reaction solvent was distilled off below 40°C under reduced pressure and added water to the residue. Reaction mass was then acidified with aqueous mineral acid. The aqueous layer was then basified and product was extracted in ethyl acetate. Organic layer was washed with water and dried over sodium sulphate. The solvent was evaporated under reduced pressure and the residue was purified by column chromatography on silica gel using a mixture of ethyl acetate and hexane (5/95) as eluent to give 0.8g of (I) as yellow solid. Purity (by HPLC) = 98%
Example 15
Preparation of l-(3-hydroxypropyl)-5-[(2R)-({2-[2-(2, 2, 2-trifIuoroethoxy) phenoxy]-ethyl} amino) propyl]-2, 3-dihydro-lH-indole-7-carbonitriIe (XVII) A mixture of 3-[7-Cyano-5 (R)-[-2-{2-[2-(2,2,2-trifluoroethoxy)-phenoxy] ethyl} amino) propyl]-2,3-dihydro-lH-indol-l-yl}propyl benzoate (XVI) (6.0 g , 0.010 mole), methanol (30 ml) and aqueous solution of Sodium hydroxide ( 1.6 g in 8 ml of water) was stirred at ambient temperature for 6 hours. To the reaction mixture water (90ml) was added and product was extracted with ethyl acetate (90 ml). The organic layer was washed with saturated sodium bicarbonate solution followed by brine wash and dried over anhydrous sodium sulfate. The solvent was evaporated under reduced pressure to give 3.85 g of (XVII). Example 16
Preparation of l-(3-Hydroxy-propyl)-5(R)-(2-{2-[2-(2, 2, 2-trifluoro-ethoxy)- phenoxy]-ethylamino}-propyl)-2, 3-dihydro-lH-indol-7-carboxylic acid amide (I) (Silodosin)
To a solution of l-(3-hydroxypropyl)-5(R)-[2-({2-[2-(2,2,2-trifluoroethoxy)phenoxy]- ethyl}amino)propyl]-2,3-dihydro-lH-indole-7-carbonitrile (XVII) (6.0 g , 0.013 mole) in dimethylsulfoxide (75 ml) was added 5 N sodium hydroxide solution (4.5 ml). To this reaction mixture, 30 % hydrogen peroxide (2.63 ml) was added slowly below 25°C. Reaction mixture was stirred at ambient temperature for 6 hours. Aqueous solution of sodium sulfite (2.1 in 150 ml water) was added to the reaction mixture. The reaction mixture was extracted with ethyl acetate. The combined ethyl acetate layer was extracted 2N hydrochloric acid. The aqueous layer was neutralized with sodium bicarbonate and extracted the product in ethyl acetate. The organic layer was washed with saturated sodium bicarbonate solution followed by brine wash and dried over anhydrous sodium sulfate. The solvent was evaporated under reduced pressure, and the residue was dissolved in ethyl acetate. The resulting solution was cooled to 5°C and filtered to get 4.51 g of (I) as solid.
.........................................................
WO2012147019
The present invention provides a process for the preparation of Silodosin of formula (I). More particularly, the present invention provides the process for preparation of tartrate salt of 3-[7-cyano-5[(2R)-2-({2-[2-(2,2,2- trifluoroethoxy)phenoxy ] ethyl } amino)propyl] -2, 3 -dihydro- 1 H-indol- 1 -y 1 } propyl benzoate of formula (IV), which is a precursor in the preparation of Silodosin.

Background of the Invention:
A compound of 3-[7-cyano-5[(2R)-2-({2-[2-(2,2,2-trifluoroethoxy) phenoxy] ethyl}amino)propyl]-2,3-dihydro-lH-indol-l-yl}propyl benzoate (IV) is a key intermediate for preparation of Silodosin. The chemical name of Silodosin is l-(3- hydroxypropyl)-5-[(2R)-2-({2-[2-(2,2,2-trifluoroethoxy)phenoxy]ethyl} amino) propyl]-2,3-dihydro-lH-indole-7-carboxamide and structurally represented as
(IV)

(I)
U.S.Pat. No. 5,387,603 discloses Silodosin as therapeutic agents for the treatment of dysuria, urinary disturbance associated with benign prostatic hyperplasia.
U.S.Pat. No. 6,310,086 discloses a process for preparing a Silodosin analogue compound from reaction of (R)-3-{5-(2-aminopropyl)-7-cyano-2,3- dihydro- 1 H-indol- 1 -yl jpropylbenzoate with 2-(2-Ethoxyphenoxy)ethyl methane sulfonate and finally isolated as residue and purified by column chromatography on silicagel. The said literature process has certain drawbacks like use of column chromatography.
U.S.Pat. No. 7,834,193 (IN 3178/DELNP/2007) discloses the process for preparation of monooxalate salt of 3-{7-cyano-5-[(2R)-2-({2-[2-(2,2,2- trifluoroethoxy)phenoxy ] ethyl } amino)propyl] -2, 3 -dihydro- 1 H-indol- 1 -y 1 } propyl benzoate (IV). This patent specifically discloses the preparation of monooxalate salt of formula (IV) helps to remove N,N-dialkyl impurity to certain extend. CN 101993405 A discloses the reaction of (R)-5-(2-aminopropyl)-l-(3-(4- fluorobenzoyloxy)propyl)-7-cyanoindoline with 2-(2-(2,2,2-trifluoroethoxy) phenoxy)ethyl methane sulfonate followed by oxalic acid salt preparation.
The main drawback in the prior art process, the formation of N,N-dialkyl impurity compound of formula (VI), as disclosed in detailed description, in the preparation of Silodosin, during condensation of compound of formula (II) with compound of formula (III), the impurity which is not removable by crystallization method or precipitation technique and column chromatography purification is not suitable for commercial purpose. So considering the commercial importance of Silodosin, the present invention focus on the preparation of pure Silodosin, and surprisingly found that the isolation of formula (IV) as tartrate salt helps to prepare Silodosin having less than 0.2 % of N,N dialkyl impurity and with good yield. None of the prior arts teaches or motivates isolation of tartaric acid addition salt of formula (IV). The preparation of Silodosin from tartrate salt of 3-{7-cyano-5-[(2R)- 2-({2-[2-(2,2,2-trifluoroethoxy)phenoxy] ethyl}amino)propyl]-2,3-dihydro-lH- indol-l-yl} propyl benzoate (IV) or its freebase of the present invention has purity of greater than 99.6 %.
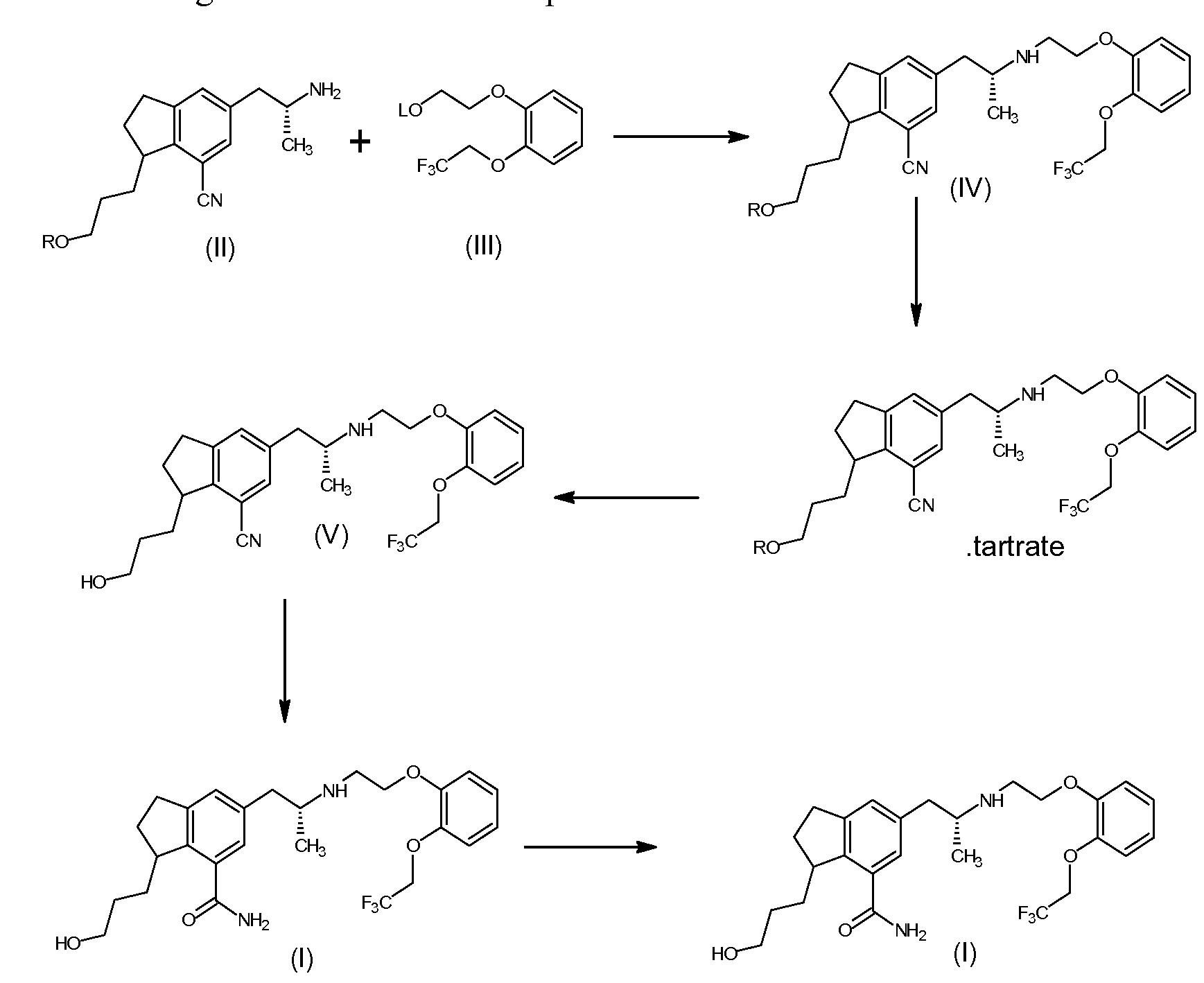
Example 3
Preparation of l-(3-hydroxypropyl)-5-[(2R)-2-({2-[2-(2,2,2-trifluoroethoxy) phenoxy] ethyl-} amino) propyl]-2,3-dihydro-lH-indole-7-carboxamide (Silodosin)
Method A: The compound of l-(3-hydroxypropyl)-5-[(2R)-2-({2-[2-(2,2,2- trifluoroethoxy)phenoxy] ethyl- } amino)propyl] -2,3 -dihydro- 1 H-indole-7- carbonitrile of formula (V) in dimethylsulfoxide was treated with 48% hydrogen peroxide and 20% sodium hydroxide solution and stirred at room temperature till completion of reaction. After completion of reaction, reaction mass quenched with 5% sodium bisulphite solution and ethylacetate was added over it. The ethylacetate layer was separated and treated with 20 % aqueous hydrochloric acid. The aqueous layer separated, neutralized with sodium bicarbonate solution and extracted with ethylacetate. The separated organic layer was washed with 10% sodium bicarbonate solution, brine solution and dried under vacuum. The organic layer distilled upto residue under vacuum at 50-55°C. The obtained residue was crystallized in ethylacetate.
Method B: To the tartrate salt of 3-[7-cyano-5[(2R)-2-({2-[2-(2,2,2- trifluoroethoxy) phenoxy] ethyl}amino)propyl]-2,3-dihydro-lH-indol-l-yl}propyl benzoate (IV) (100 grams) in methanol, aqueous potassium hydroxide solution (38.38 grams) was added and stirred at room temperature till reaction completion. After completion of reaction, DM water and dichloromethane was added over it under stirring. Organic layer separated, washed with brine solution distilled under vacuum upto less than 1 volume. To the solution, dimethyl sulphoxide, 20% sodium hydroxide and hydrogen peroxide was added and stirred till completion of reaction. After completion of reaction, water containing sodium bisulfite was added to the reaction mass. The pH of the reaction mixture adjusted to about 8.5 using 10% sodium hydroxide and extracted in dichloromethane twice, washed with water, dried and concentrated upto 1-2 volume under vacuum. To the obtained solution, toluene was added over it at room temperature under stirring. The reaction mixture maintained for complete solid formation, filtered and dried under vacuum. Yield 58 grams. Example 4
Purification of Silodosin:
Method A: To the mixture of toluene and acetonitrile solvent, Silodosin was added over it and heated to 50° - 55 °C for complete dissolution. The reaction mass gradually cooled to room temperature and maintained for completion of solid formation. The obtained solid is filtered, washed with toluene and dried under vacuum. Method B: To the mixture of ethyl acetate and toluene solvent, Silodosin was added over it and heated to 60° - 65 °C for complete dissolution. The reaction mass gradually cooled to room temperature and maintained for completion of solid formation. The obtained solid is filtered, washed with toluene and dried under vacuum.
//////////////////////////////////////////
CN101993407
silodosin for selective inhibition of urethral smooth muscle contraction and reduce the pressure within the urethra, but no significant impact on blood pressure, for the treatment of benign prostatic hyperplasia. At present, the method of synthesis Silodosin many reports, but the lack of high yield method for industrial production.
JP200199956 reported that benzoic acid as a starting material, 1_ (3_ benzoyloxy-propyl) indoline hydrochloride (structural formula (1), R is a hydrogen atom) in 60% yield, then through the multi-step reaction was further prepared silodosin intermediate 1- (3-benzoyloxy-propyl) -5- (2-nitro-propyl) -7-cyano-indoline (structural formula VIII ), the total yield is low, and only 20 percent. Compound (VIII) with potassium carbonate, the reaction of hydrogen peroxide to yield compound (IX), impurities, and purified by column chromatography to be not suitable for industrial production. Compound (IX) under catalysis of molybdenum oxide, and L- (S) - benzyl glycyl alcohol asymmetric reactions, protecting groups may be due to steric hindrance is small, low chiral induction, is 3.8: I.


Silodosin Preparation: 12 Example
Example 11 to give 8 g solid, dissolved in DMSO 100ml, was added 5mol / L NaOH 12ml, 18 ~ 20 ° C was added dropwise slowly with 30% H2027 grams, then 30 ° C, the reaction ended 4h. Extracted with ethyl acetate, the combined organic layer was washed 2N HCl and then the organic layer, the aqueous layer was neutralized with sodium hydroxide, and then extracted with ethyl acetate, washed with saturated sodium bicarbonate, dried over anhydrous sodium sulfate, and evaporated concentrated and then dissolved in ethyl acetate, natural cooling crystallization, filtration, drying 5 g (87%), purity> 99%.
Mp 105 ~ 108 ° C
[a] 20d = -16.2 C = I, MeOH
1NMR spectrum (DMS0-d6): δ ppm 0.9-1.0 (3H, d), 1.5-1.6 (1H, s), 1.6-1.7 (2H, m),
2.3-2.4 (1H, dd), 2.6-2.7 (1H, dd), 2.8-3.0 (5H, m), 3.1-3.2 (2H, m), 3.3-3.4 (2H, m),
3.4-3.5 (2H, t), 4.0-4.1 (2H, t), 4.2-4.3 (1H, s), 4.6-4.8 (2H, t), 6.9-7.15 (6H, m),
7.2-7.3 (1H, s), 7.5-7.6 (1H, s)
...........................................................
see
Silodosin is (I) (formula 1, claim 1, page 31).
Process for the prepartion of pure polymorphic gamma form of silodosin - comprising dissolving any polymorphic form of silodosin in a solvent and seeding gamma form of silodosin.
Crude (I) (50 g) was dissolved in methanol, filtered and solvent was distilled under vacuum. The residue was dissolved in isopropanol at 50 degreeC, cooled and seed of (I) gamma form was added and further cooled and cyclohexane (500 mL) was added, solid was filtered, washed and dried to obtain pure polymorphic form gamma of (I) having a toluene content of 12 ppm (example 10, pages 29-30).
A process for the preparation of silodosin and/or its salt is claimed, comprising the reaction of 3-[5-((2R)-2-aminopropyl)-7-cyano-2,3-dihydro-1H-indol-1-yl]propyl benzoate(2R,3R)-monotartrate with 2-[2-(2,2,2-trifluoroethoxy)phenoxy]ethyl methanesulfonate to form a cyano benzyloxy intermediate, followed by hydrolysis to form a cyano hydroxy intermediate, which is then reacted with tartaric acid and hydrolyzed in the presence of an oxidizing agent to obtain the product. An alternate method of preparation of silodosin comprising the hydrolyses of tartrate salt of cyano hydroxy intermediate in the presence of an oxidizing agent, pure polymorphic form gamma of silodosin, and the cyano hydroxy intermediate are also claimed. Further processes for the prepartion of the pure polymorphic form gamma of silodosin are claimed, wherein the process involves the dissolution of of any polymorphic form of silodosin in a solvent by heating at 30-100 degree C, cooling before and after seeding with gamma form of silodosin, adding an antisolven, isolating the polymorph and optionally micronizing.
The present invention provides an improved and efficient process for the preparation of

It acts as an selective ai -adrenoceptor antagonist and is useful in the symptomatic treatment of benign prostatic hyperplasia (BPH). Chemically it is known as l-(3-hydroxypropyl)-5-[(2R)- ( { 2-[2-[2-(2,2,2-trifluoroethoxy)phenoxy]ethylamino) propyl] indoline-7-carboxamide.
Silodosin and its pharmaceutically acceptable salts are first disclosed in US patent 5,387,603. Synthetic approach for the production of silodosin, is described in patent '603 can be represented as shown below in scheme 1.
l

Scheme 1
As represented in scheme 1, silodosin is prepared by the reaction of l-acetyl-5-(2r aminopropyl)indoline-7-carbonitrile with 2-[2-(2,2,2-trifiuoroethoxy)phenoxy] ethyl methanesulfonate in the presence of sodium bicarbonate in ethanol to give l-acetyl-5-[2-[2-[2-(2,2,2-trifluoroethoxy)phenoxy]ethylamino]propyl]indoline-7-carbonitrile, which upon reaction with di-tert-butyldicarbonate in methylene chloride produces protected acetyl indoline carbonitrile compound. Further deacetylation with sodium hydroxide in ethanol followed by treatment with acetic acid provides protected indoline carbonitrile compound, which upon hydrolysis using dimethyl sulfoxide, 30% hydrogen peroxide, sodium hydroxide and acetic acid gives protected indoline carboxamide, which upon further reaction with 2-tert-butyldimethylsiloxy)ethyl-4-nitrobenzene sulfonate in the presence of cis-dicyclohexano-18 crown-6 and potassium carbonate in dioxane gives protected (tert-butyl-dimethylsiloxy) ethyl indoline carbonitrile. Further treatment with tetrabutylammonium fluoride in tetrahydrofuran produces N-boc protected hydroxy deprotected propyl indoline carbonitrile, which under goes facile deprotection of boc group upon treatment with trifluoroacetic acid, in methylene chloride to yield silodosin. The complete process is very complex, make use of pyrophoric reagents
which are very difficult to handle in large scale and have many extra steps involving protection and depfotection. Further in US patent '603, concrete detail of preparation and purification of silodosin have not been reported. Furthermore, isolated silodosin is characterized using IR, NMR and specific rotation but the patent is silent on product appearance and crystalline nature. There are several processes known for the preparation of silodosin and its intermediates viz; in JP 4634560; JP 4921646; JP-2006- 188470; WO2011/124704 and WO2011/101864. In most of the inventions, silodosin is prepared by following reaction as shown in scheme 2. Major disadvantages of these processes are the formation of N,N dialkyl impurity, and other impurities which forms during the condensation of 3-[5-((2/?)-2-aminopropyl)-7-cyano-2,3-dihydro-lH-indol-l-yl]propyl benzoate or its salts like monotartrate with 2-[2-(2,2,2-trifluoroethoxy)phenoxy] ethyl methanesulfonate. N,N dialkyl impurity forms in about 12-15% and may form due to reaction of one molecule of benzoate compound with two molecules of methanesulfonate compound. Removal of this impurity is not possible by simple purification

wherein R is benzoyl, benzyl, tetrahydropyranyl, 2-trimethylsilylethyl, dinitrophenyl, diphenyl methyl and the like
Scheme 2
US patent 7,834,193 discloses a process for preparation of silodosin with similar condensation of 3-[5-((2R)-2-arriinopropyl)-7-cyano-2,3-dihydro-lH-indol-l-yl]pfopyl benzoate or its salts like monotartrate with 2-[2-(2,2,2-trifluoroethoxy)phenoxy]ethyl methanesulfonate, but 3-{7-cyano-5-[(2R)-2-({2-(2,2,2-trifluoroethoxy)-phenoxy]ethyl}amino)propyl)-2,3-dihydro-lH-indol-l-yl)-propylbenzoate is purified by preparing monooxalate salt as shown below in
scheme 3. This patent specifically prepares monooxalate salt of 3- {7-cyano-5-[(2R)-2-({ 2- (2,2,2-trifluoroethoxy)-phenoxy]ethyl }amino)propyl)-2,3-dihydro-lH-indol-l-yl)-propyl benzoate to remove N,N÷dialkyl impurity, but impurity has not been removed completely, only a certain % of it, has been removed.

Scheme 3
In PCT publication WO2012/131710, preparation of silodosin is described wherein improved processes for preparation of 3-[5-((2R)-2-aminopropyl)-7-cyano-2,3-dihydro-lH-indol-l- yl]propyl benzoate have been disclosed which is then converted to silodosin by condensation with 2-[2-(2,2,2-trifluoroethoxy)phenoxy]ethyl methanesulfonate. In exemplified process, 3-[5- ((2R)-2-aminopropyl)-7-cyano-2,3-dihydro-lH-indol-l-yl]propyl benzoate is condensed with 2-[2-(2,2,2-trifluoroethoxy)phenoxy]ethyl methanesulfonate and the resulting benzoate compound is hydrolyzed to give l-(3-hydroxy propyl)-5-[(2R)-2-({ 2-[2,2,2-trifluoroethoxy) phenoxy] ethyl }amino)propyr]-2,3-dihydro-lH-indol-7-carbonitrile.The carbonitrile compound is treated with oxalic acid to prepare its oxalate salt having purity greater than 99%, which is then hydrolyzed using a base to prepare free carbonitrile compound having purity greater than 99%, but this patent is silent about N, N- dialkyl impurity or its removal.
In PCT publication WO2012/147019, preparation of silodosin using 3-{ 7-cyano-5-[(2R)-2-({2- (2,2,2-trifluoroethoxy)-phenoxy]ethyl}amino)propyl)-2,3-dihydro-lH-indol-l-yl)-propyl benzoate tartrate salt has been described as shown below in scheme 4.

Scheme 4
One other PCT publication WO2012/147107 describes preparation of silodosin by preparing hydrochloride and acetic acid salts of l-(3-hydroxypropyl)-5-[(2R)-2-({2-[2,2,2-trifluoroethoxy) phenoxy] ethyl }amino)propyl]-2,3-dihydro-lH-indol-7-carbonitrile to remove N,N dialkyl impurity. It has been observed that in exemplified process, wherein hydroxy compound namely l-(3-hydroxy propyl)-5-[(2R)-2-({2-[2,2,2-trifluoroethoxy) phenoxy] ethyl }amino)propyl]-2,3-dihydro-lH-indol-7-carbonitrile is purified by preparing its acetate salt to, remove the impurities but still N, N-dialkyl impurity remains in an amount of 0.6%, which is difficult to remove in next stage or require extra purifications.
Beside to use highly pure silodosin, use of a pure polymorphic form of API is an essential requirement of drug formulation, these both aspects when address jointly, and obtained silodosin can be converted to pure polymorph then only a complete solution of prior art problems can be achieved. Apart from above mentioned process patents/publications which aimed to prepare the pure silodosin, there are exist some polymorph patents/publications which also aims to prepare pure polymorphic form of silodosin.
Polymorphism is considered as one of the- most important solid-state property of drug substance, since different polymorph have different physiochemical and biological properties and in pharmaceutical chemistry it is often desired to obtain one particular form that is biologically active and also offers ease of handling during formulation. The available literature references related to polymorph of silodosin are incorporated herein.
Japanese patent 3331048 (publication No.H07-330726), discloses a process for purification of silodosin wherein silodosin is dissolved in ethyl acetate, dried over anhydrous magnesium sulfate, solvent is distilled off and again dissolved in ethyl acetate at 70°C and crystallizes below room temperature. The resulting product is characterized by melting point, IR, NMR and specific rotation. Here also disclosure is silent about polymorphic form of product.
US patent publication US2006/0142374A1 (equivalent European patent EP1541554B 1) discloses polymorphic forms of silodosin including three crystalline polymorphic form of silodosin which are named as alpha (a), beta (β) and gamma (γ) and one amorphous form. These polymorphic forms have been characterized by X-ray powder diffraction pattern. In the patent publication, processes for the preparation of all these three crystalline forms have been disclosed. In. a given process, form alpha is prepared by dissolving crude silodosin in appropriate amount of ethyl acetate, ethyl formate, acetone, methyl ethyl ketone, acetonitrile, tetrahydrofuran or mixture of acetone and acetonitrile (1: 1), preferably ethyl acetate under heating, allowing to stand at room temperature to precipitate the crystal gradually. Similarly, form beta is prepared by dissolving crude silodosin in appropriate amount of methanol under heating, adding petroleum ether as a anti-solvent, crystal precipitation is ensured using vigorous stirring.
In a second process, to prepare the form beta, crude silodosin is dissolved in ethanol or 1-propanol and the reaction mass is cooled quickly. The crystalline form gamma is prepared by dissolving crude silodosin in appropriate amount of toluene or a mixture of acetonitrile and toluene (1:4) or ethyl acetate and toluene (1: 19), preferably in toluene, under heating, cooling to room temperature and allowing to precipitate gradually upon standing. In a second process to prepare form gamma, crude silodosin is dissolved in 2-propanol and the crystals are precipitated by adding an appropriate amount of toluene. In spite of disclosing three crystalline polymorphic forms, the patent publication prefers preparation and use of form alpha by highlighting the problems faced for preparation and use of other forms. It is disclosed that crystal form beta has manufacturing difficulties at industrial scale since precipitation occurs only when the nonpolar antisolvent is added to warm solution which leads to inconsistency in quality of crystals.
With the second process for preparation of form beta, desired level of yield and purity has not been achieved. Further, according to this publication, preparation of gamma form involves use of toluene which can not be removed completely from final product, because of its high boiling point and raises the problem of residual solvent. In the case of toluene, a class 2 solvent, its limits should not be more than 890 ppm. In the exemplified process, toluene content has not been disclosed, which clearly reflects that product was not suitable for pharmaceutical composition having problem of high residual content of toluene. Furthermore patent publication also states that all the three crystal forms donot have any difference in hygroscopicity and stabilities.
Thereafter, several patents/publications disclose preparation of polymorphic forms alpha and beta. For example a PCT publication WO2012/147107 discloses a process for preparation of beta form using isopropyl acetate and methyl isobutylketone. In another PCT publication WO2012/077138, preparation of alpha and beta forms are disclosed using various solvent , system. Similarly, in a Chinese patent CN102010359, crystalline form beta is prepared by dissolving the crude silodosin in alcoholic solvent by heating and the product is crystallized by cooling or by adding an antisolvent such as ketone or ether.
European patent EP2474529 discloses new polymorphic forms delta (δ) and eta (ε) of silodosin by using a solvent (tetrahydrofuran) and antisolvent (n-heptane, n-hexane, cyclohexane, tert butylmethyl ether).Further it discloses conversion of delta form to beta form by just heating the delta form at a particular temperature. The form delta can also be transformed into form eta by. slurrying in aqueous methanol. One new crystalline form designated as delta has also been disclosed in a Chinese patent publication CN102229558. An Indian patent application 478/MUM/2010, also discloses a new polymorphic form Zy-S which is prepared by using solvent such as esters, aromatic hydrocarbons, ketones, and alcohols.
All the above disclosures are silent about the preparation of gamma form of silodosin and only available disclosure reports that gamma form have problem of residual solvent, as impurity and is not suitable for pharmaceutical compositions.
Method C: l-(3-HydroxypropyI)-5-[(2R)-2-({2-[2,2,2-trifIuoroethoxy)phenoxy] ethyl} amino) propyl]-2,3-dihydro-lH-indol-7-carbonitrile tartrate (lOg) dissolved in dimethylsulfoxide (120 ml) and to this solution, was added 5 mol/L aqueous sodium hydroxide solution (15ml). To the reaction mixture, 30% hydrogen peroxide (5ml) was added and keeping the temperature below 25°C. The reaction mixture was stirred at 20-25°C, for 5 hours. To the reaction mixture, sodium sulfite (5g) dissolved in water (100ml) was added slowly. The reaction mixture was extracted with ethyl acetate (1x200ml) and ethyl acetate layer was concentrated under reduced pressure. The resulting product was dissolved in methanol and clear solution was filtered through micron filter paper of size 0.22 micron two times and filtrate was concentrated.The resulting compound was dissolved in toliiene (70ml) and isopropyl alcohol (7ml) at 50-55°C and the solution was cooled to 20-25°C, cyclohexane was added and stirred for further 4 hours, filtered and dried to give title compound having purity 99.86% and N,N-dialkyl impurity not detected by HPLC. Example 5: Preparation of pure Polymorphic Form Gamma (γ) of Silodosin
Silodosin (15g) having toluene content 1872 ppm, was micronized under air pressure. The micronized product was dried under vacuum at 55°C-60°C for 23.0 hours to afford pure polymorphic form gamma of silodosin having toluene content 460 ppm.
Example 6: Preparation of pure Polymorphic Form Gamma (γ) of Silodosin
Silodosin [having toluene content 1327 ppm] was micronized under air pressure. The micronized product was dried under vacuum at 55°C-60°C for 16 hours to afford pure polymorphic form gamma of silodosin having toluene content 350 ppm.
Example 7: Preparation of pure Polymorphic Form Gamma (γ) of Silodosin
Silodosin crude (3.0g) was dissolved in isopropanol (12ml) at 50°C and reaction mass was cooled to 35°C and seed of silodosin gamma form (O.lg) was added. Thereafter reaction mass was again cooled to 15-20°C and cyclohexane (30ml) was added to the reaction mass and stirred for further 0.5 hour. The resulting solid, thus obtained, was filtered, washed with cyclohexane and dried to afford pure polymorphic form gamma of silodosin having toluene content 34 ppm.
References
- "Drugs.com, Watson Announces Silodosin NDA Accepted for Filing by FDA for the Treatment of Benign Prostatic Hyperplasia". Retrieved 2008-02-13.
- European Medicines Agency: Assessment report for Silodyx
- Schubert-Zsilavecz, M, Wurglics, M, Neue Arzneimittel 2008/2009
- Kobayashi K, Masumori N, Kato R, Hisasue S, Furuya R, Tsukamoto T. (December 2009). "Orgasm is preserved regardless of ejaculatory dysfunction with selective alpha1A-blocker administration.". Int J Impot Res. 21 (5): 306–10. doi:10.1038/ijir.2009.27. PMC 2834370. PMID 19536124.
External links
a1a-Adrenoceptor antagonist. Prepn: M. Kitazawa et al., EP 600675; eidem, US 5387603 (1994, 1995 both to Kissei).PRODUCT PATENT
Adrenoceptor binding study: K. Shibata et al., Mol. Pharmacol. 48, 250 (1995); and tissue selectivity: S. Murata et al., J. Urol. 164, 578 (2000).
Pharmacology: K. Akiyama et al., Pharmacology 64, 140 (2002).
Series of articles on pharmacology, pharmacokinetcs and toxicology: Yakugaku Zasshi 126, 187-263 (2006).
Review of development and therapeutic potential: F. Kamali, Curr. Opin. Cent. Peripher. Nerv. Syst. Invest. Drugs 1, 248-252 (1999)
| CN101993405A * | Aug 27, 2009 | Mar 30, 2011 | 浙江华海药业股份有限公司;上海医药工业研究院 | Indoline derivative as well as preparation method and application thereof |
| JP2006188470A * | Title not available | |||
| US7834193 * | Apr 16, 2007 | Nov 16, 2010 | Kissei Pharmaceutical Co., Ltd. | industrial production of silodosin (for treating dysuria associated with benign prostatic hyperplasia) via mixing 3-{7-cyano-5-[(2R)-2-({2-[2-(2,2,2-trifluoroethoxy)-phenoxy]ethyl}amino]propyl]-2,3-dihydro-1H-indol-1-yl}-propyl benzoate and oxalic acid, nitrilizing, hydrolyzing |
| WO2011030356A2 * | Sep 13, 2010 | Mar 17, 2011 | Sandoz Ag | Process for the preparation of indoline derivatives and their intermediates thereof |
| WO2011124704A1 * | Apr 8, 2011 | Oct 13, 2011 | Ratiopharm Gmbh | Process for preparing an intermediate for silodosin |
| WO2012131710A2 * | Mar 27, 2012 | Oct 4, 2012 | Panacea Biotec Ltd | Novel process for the synthesis of indoline derivatives |
| JP2006188470A * | Title not available |
| Patent | Submitted | Granted |
|---|---|---|
| Combination therapy for the treatment of benign prostatic hyperplasia [US6410554] | 2002-06-25 | |
| Indoline compound and process for producing the same [US7834193] | 2007-08-23 | 2010-11-16 |
| Agents and crystals for improving excretory potency of urinary bladder [US8252814] | 2009-10-22 | 2012-08-28 |
| METHODS FOR TREATING BENIGN PROSTATIC HYPERPLASIA [US2011319464] | 2011-12-29 | |
| PREVENTIVE AND/OR THERAPEUTIC AGENT FOR URINE COLLECTION DISORDER ACCOMPANYING LOWER URINARY TRACT OBSTRUCTION [US2009227651] | 2009-09-10 | |
| PREVENTIVE AND/OR THERAPEUTIC AGENT FOR URINE COLLECTION DISORDER ACCOMPANYING LOWER URINARY TRACT OBSTRUCTION [US2010137399] | 2010-06-03 | |
| Agents for improving excretory potency of urinary bladder [US2004116457] | 2004-06-17 | |
| Medicinal Composition for Prevention of Transition to Operative Treatment for Prostatic Hypertrophy [US2008090893] | 2008-04-17 | |
| METHODS FOR TREATING BENIGN PROSTATIC HYPERPLASIA [US2008242717] | 2008-10-02 | |
| Agents and crystals for improving excretory potency of urinary bladder [US2006281725] | 2006-12-14 |
Capromorelin Tartrate is the Tartrate salt of Capromorelin, also known as CP-424,391, which is an orally active, potent growth hormone secretagogue receptor (GHSR) agonist, with Ki of 7 nM for hGHS-R1a. Capromorelin Tartrate
ReplyDeleteAtorva 80 MG Tablet is used to reduce the levels of bad cholesterol and triglycerides in the blood. Along with taking this medicine, a change in diet and regular exercise is recommended. The medicine helps in reducing the long-term cardiovascular risks such as stroke, heart attacks, chest pain, etc.
ReplyDelete