TELMISARTAN PART 3/3

CONT.........................
PAPER
Journal of Organic Chemistry (2015), 80(3), 1915-1919
J. Org. Chem., 2015, 80 (3), pp 1915–1919
DOI: 10.1021/jo5025333

A direct and efficient total synthesis has been developed for telmisartan, a widely prescribed treatment for hypertension. This approach brings together two functionalized benzimidazoles using a high-yielding Suzuki reaction that can be catalyzed by either a homogeneous palladium source or graphene-supported palladium nanoparticles. The ability to perform the cross-coupling reaction was facilitated by the regio-controlled preparation of the 2-bromo-1-methylbenzimidazole precursor. This convergent approach provides telmisartan in an overall yield of 72% while circumventing many issues associated with previously reported processes.
........................
.
PAPER
International Journal of Research in Pharmaceutical and Biomedical Sciences (2013), 4(1), 293-295
telmisartan1. [Yield 87%, Purity 99.97% by HPLC.M.P. 260 – 262°C, Sulphated ash < 0.01%].
1H NMR (DMSO-d6): δ 0.98-1.03 (t,3H), 1.73- 1.86 (m, 2H), 2.5 - 2.63 (s, 3H), 2.90-2.95 (s, 2H),3.82 (s, 3H), 5.62 (s, 2H), 7.16-7.34 (m,7H), 7.40-7.59 (m,4H), 7.68-7.70 (m, 3H), 12.86 (s, 1H).
M/Z: 515.50 [M + H]+
.............................
.
PATENT
PATENT
WO 2014027280
Scheme 1 given below: Formula .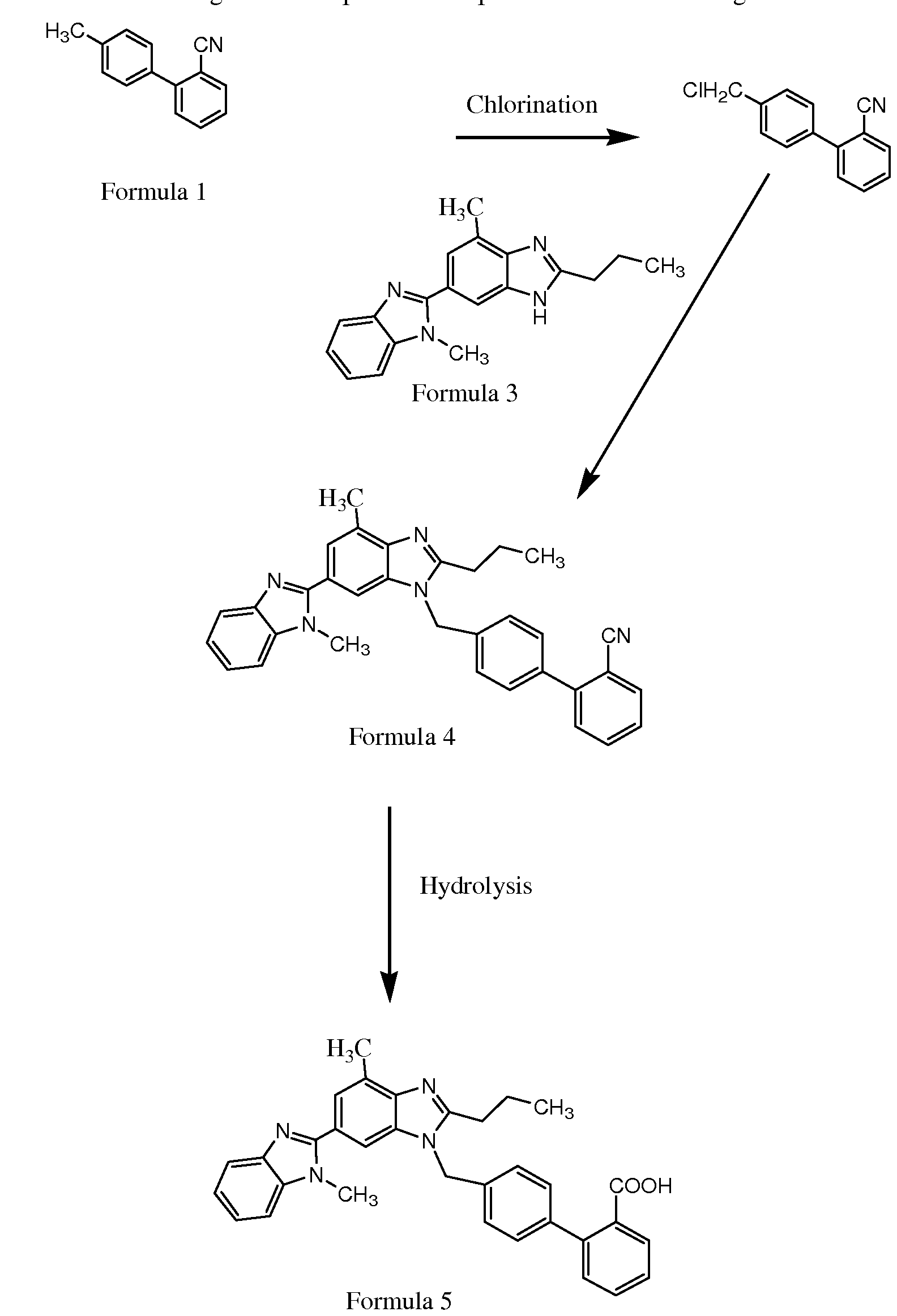
The lower aqueous layer was separated and the upper organic layer was washed with 300 ml water. The lower aqueous layer was separated. To the organic layer containing 4-chloromethyl-2'-cyanobiphenyl, the compound 2-n-propyl-4-methyl-6-(l'- methylbenzimidazol-2'-yl)benzimidazole was added, followed by a solution of 40 gm sodium hydroxide in 300 ml water.
The reaction mass was stirred for 10 minutes and 10 gm of tetrabutyl ammonium hydrogen sulphate was added. The reaction mass was heated to 80 UC and maintained at 80 to 85 °C for 4 hours. The completion of the reaction was monitored by TLC using mobile phase chloroform: methanol (9: 1). After completion of reaction, the lower aqueous layer was separated.
The solvent was distilled out till mass temperature 120 °C and final traces were removed completely under vacuum. To the residual mass, 50 ml of n-butanol was added and the solvent distilled out under vacuum below 100 °C to remove all traces of methyl isobutyl ketone. The residue was dissolved in 750 ml of n-butanol and 83 gm sodium hydroxide added. The reaction mass was heated to reflux and maintained for 24 hours at 123 to 126 °C. The completion of the reaction was monitored by TLC using mobile phase chloroform: methanol (9: 1). The solvent was distilled out at atmospheric pressure till the mass temperature reached 140 C.
The residual mass was cooled to 100 °C and 300 ml water was added. The solvent was distilled out azeotropically till the mass temperature reached 120 °C. To the reaction mass 750 ml of water was added, the solution warmed to 80 °C. The pH of the reaction mass was adjusted to 8.0 with hydrochloric acid. Finally the pH was adjusted to 6.0 with acetic acid, and the reaction mass maintained at 80 to 85 °C for one hour. The product obtained was filtered, washed with water and dried to yield 120 gm of 4'-[2-n-propyl-4-methyl-6-(l- methylbenzimidazol-2-yl)benzimidazol-l-ylmethyl]biphenyl-2-carboxylic acid, which can be purified as per the procedure described mentioned in Example 5.
Example 2: 4-chloromethyl-2 '-cyanobiphenyl In a 1 litre reaction flask 400 ml of methylene chloride was added followed by 100 gm of 2- cyano-4' -methyl biphenyl. The reaction mass was stirred to get a clear solution and cooled to 20 °C. Chlorine gas was sparged into the reaction mass for a period of 15 hours at 20 to 25 °C till completion of the reaction. The reaction was monitored by TLC using mobile phase n- hexane: ethyl acetate (8:2). The excess chlorine from the reaction mass was removed by flushing with nitrogen. The solvent was distilled out completely by distillation at atmospheric pressure and removal of the final traces under vacuum. To the residual mass, 400 ml of n- heptane was added. The reaction mass was stirred and warmed to 60 °C. The clear solution obtained was cooled to 10 °C and the product precipitated was filtered, washed with n-heptane and dried. Further crystallization with n-heptane yielded 80 gm of pure 4-chloromethyl-2'- cyanobiphenyl. C 73.87%, H 4.41%, N 6.19%; m/z 192.25; 1H NMR DMSO d6400 Mhz : 5ppm 4.84 (s, 2H) 7.32 - 7.66 (aromatic 8H).
Example 3: 2-cyano-4,-(2,,-n-propyl-4,,-methyl-6,,-{V"-methylbenzim ylmethyl) biphenyl In a 2 litre reaction flask 500 ml of methyl isobutyl ketone was added followed by 100 gm of 2-n-propyl-4-methyl-6-( -methylbenzimidazol-2'-yl)benzimidazole. The reaction mass was stirred and a solution of 40 gm sodium hydroxide in 300 ml water was added. To this solution, 10 gm tetra butyl ammonium hydrogen sulphate and 80 gm of 4-chloromethyl-2'- cyanobiphenyl was added. The reaction mass was warmed to 80 °C and maintained for 4 hours at 80 to 85 °C. The completion of the reaction was monitored by TLC using mobile phase chloroform : methanol (9:1). After completion of the reaction, the mass was cooled to 20 °C, maintained 3 hours at 15 to 20 °C. The product which precipitated out was filtered, washed with methyl isobutyl ketone, followed by water to yield 126 gm of 2-cyano-4'-(2"-n-propyl-4"-methyl- 6"-(r"-methylbenzimidazol-2"'-yl)benzimidazol-l"- ylmethyl) biphenyl, melting at 196 - 198 °C. C 80.53%, H 5.70%, N 14.20%; m/z = 496.64 lH NMR DMSO d6 400 Mhz : 5ppm 0.96 - 0.99 (t, 3H) 1.75 - 1.84 (m, 2H) 2.62 (s, 3H) 2.89 - 2.93 (t, 2H) 3.80 (s, 3H) 5.67 (s, 2H) 7.18 - 7.92 (m, 14H)
Example 4: 4'-[2-n^ropyl-4-methyl-6-(l-methylbenzi idazol-2-yl)benzi idazol-ylmethyl]bipheny carboxylic acid 126 gm of 2-cyano-4'-(2"-n-propyl-4"-methyl-6"-(l "'-methylbenzimidazol-2"'-yl) benzimidazol-1"- ylmethyl) biphenyl was dissolved in 750 ml of n-butanol and 83 gm sodium hydroxide added. The reaction mass was heated to reflux and maintained for 15 hours at 123 to 126 °C. The completion of the reaction was monitored by TLC using mobile phase chloroform: methanol (9: 1). The solvent was distilled out at atmospheric pressure till the mass temperature reached 140 °C. The residual mass was cooled to 100 °C and 300 ml water was added. The solvent was distilled out azeotropically till the mass temperature reached 120 °C. To the reaction mass 750 ml of water was added, the solution warmed to 80 °C. The pH of the reaction mass was adjusted to 8.0 with hydrochloric acid. Finally the pH was adjusted to 6.0 with acetic acid, and the reaction mass maintained at 80 to 85 °C for one hour. The product obtained was filtered, washed with water and dried to yield 120 gm of 4'-[2-n-propyl-4-methyl-6-(l- methylbenzimidazol-2-yl)benzimidazol-l-ylmethyl]biphenyl-2-carboxylic acid.
Example 5: Purification of 4'-[2-n^ropyl-4-methyl-6-(l-methylbenzimidazol-2-yl)benzimidazol-l- ylmethyl]biphenyl-2-carboxytic acid In a 3 litre reaction flask, 1000 ml of methanol was added followed by the addition of 120 gm of 4'-[2-n-propyl-4-methyl-6-(l-methylbenzimidazol-2-yl)benzimidazol-l-ylmethyl]biphenyl- 2-carboxylic acid obtained by procedure described in Example 4. The solution was warmed to 50 °C and pH adjusted to 10.0 to 10.5 with 100 ml of a 10% methanolic potassium hydroxide solution. The reaction mass became a clear solution, and 6 gm activated carbon was added. The mass was maintained at 50 to 55 °C for one hour and filtered through hyflo supercel to remove the activated carbon. The clear filtrate obtained was collected and its pH adjusted to 6.0 to 6.5 with 130 ml of acetic acid, maintaining the temperature between 50 to 55 °C. The mass was cooled to 15 °C and maintained one hour at 10 to 15 °C. The product which precipitated out was filtered, washed with 50 ml of methanol followed by 500 ml of water. The wet product was dried to yield 107 gm of 4'-[2-n-propyl-4-methyl-6-(l- methylbenzimidazol-2-yl)benzimidazol-l-ylmethyl]biphenyl-2-carboxylic acid. C 76.49%; H 5.74%, N 11.02%; m/z 515.45.; 1H NMR DMSO d6 400 Mhz : 5ppm 0.97 - 1.01 (t, 3H) 1.76 - 1.85 (m, 2H) 2.62 (s, 3H) 2.90 - 2.94 (t, 3H) 3.81 (s, 3H) 5.61 (s, 2H) 7.15 - 7.71 (14H aromatic); Melting point of purified telmisartan: 269 °C.
.....................
PAPER
Journal of Organic Chemistry (2014), 79(21), 10568-10580
J. Org. Chem., 2014, 79 (21), pp 10568–10580
DOI: 10.1021/jo501665e

On the basis of our recently reported aniline aqueous borylation, molecular diversity was achieved in a one-pot process by combining other reactions such as esterification, Suzuki–Miyaura coupling, hydrogenolysis, or Petasis borono-Mannich.
TELMISARTAN IS COMPD 9
...................
PATENT
US 20150031768
(EN)
Methods of halogenating a carbon containing compound having an sp3 C—H bond are provided. Methods of fluorinating a carbon containing compound comprising halogenation with Cl or Br followed by nucleophilic substitution with F are provided. Methods of direct oxidative C—H fluorination of a carbon containing compound having an sp3 C—H bond are provided. The halogenated products of the methods are provided.
.......................PATENT
WO 2014067237
Telmisartan Preparation: 12 Examples The title compound (III, R = COOCH 3) (52.8g, O. lmol) of Example 11 with glacial acetic acid(200ml) and concentrated hydrochloric acid (250ml) mixing, 100 ° C to react for 5 to 6 hours. Evaporated to most mixed acid, residue slowly poured into crushed ice, under ice cooling with saturated K 2 CO ^ solution to adjust the pH to neutral, solid precipitation, filtration, filtrate was washed with water, was for Mischa Tan crude, recrystallization telmisartan (40.1g), liquid purity greater than 99%.Example 13: Preparation of telmisartan of formula I compound (0.62g, leq) was added to acetonitrile (10ml). After stirring evenly, the KOH (0.14g, 1. leq) was slowly added, after stirring for 10 plus minutes, the title compound of Example 10 of the embodiment (11, R = COOCH 3) (0.5g, leq) was slowly added, stirred for 3-4 hours, TLC the reaction was complete, the direct addition of 50% ethanol (30mL), reflux The reaction for 6 hours. After completion of the reaction by TLC, recovering the organic solvent under reduced pressure, the remaining solution was added dropwise hydrochloric acid (1: 1) to neutral pH. The precipitated solid was filtered, washed with water to give crude telmisartan, telmisartan recrystallized (yield 75.1%), the liquid phase is greater than 98% purity. Chloromethyl biphenyl -2- (II, R = CN) Preparation of 4'-nitrile:
.....................
Journal of Pharmaceutical and Biomedical Analysis (2015), 108, 86-96.
....................
IN 262831/EP 1912975
............................
JP 2014201585
..........................
IN 2013KO00463/WO 2014174397
..................................
PATENT
Example 7: Telmisartan make 5.51 g telmisartan × HCl was dissolved in 50 ml of 40% acetic acid while refluxing. The brown solution was then filtered hot through 1.1 g of carbon, 2.5 ml of 40% acetic acid and washed, and at 80-90 ℃ 2.5 ml of 4N NaOH was added dropwise with stirring to light brown filtrate. Telmisartan crystallization, the suspension was diluted with 30 ml of water, and slowly cooled to ambient temperature. Telmisartan suction filtration, and washed with 50 ml of water. And dried in vacuo at 80 ℃ drying cabinet telmisartan. Yield: 4.80 g (93.3% of the theoretical yield)......................................
PATENT http://www.google.com/patents/CN102731407A?cl=en

 Example 4 Preparation of telmisartan [0031] 2-n-propyl group as shown in Formula I-4-methyl-6- (benzimidazol-2-yl-methyl 1'_) benzimidazole (30. 4g, O. 10mol), 4_ bromomethyl-biphenyl-2-carboxylic acid (43. 6g, O. 15mol), three ko amine (12. Ig, O. 15mol) and ko ni ni ether 500ml alcohol were mixed and reacted at 100 ° C for 6 inches The reaction solution was poured into ice water, acidified with dilute hydrochloric acid and slowly adjusted PH2-3, to precipitate a solid. Filtration, 70 ° C drying crude, the resulting crude product ko ko acid ester 300ml heating beating again. Filtered, 70 ° C dry. Recrystallization from DMF telmisartan of formula III as shown in 25. Ig, yield: 50%.
Example 4 Preparation of telmisartan [0031] 2-n-propyl group as shown in Formula I-4-methyl-6- (benzimidazol-2-yl-methyl 1'_) benzimidazole (30. 4g, O. 10mol), 4_ bromomethyl-biphenyl-2-carboxylic acid (43. 6g, O. 15mol), three ko amine (12. Ig, O. 15mol) and ko ni ni ether 500ml alcohol were mixed and reacted at 100 ° C for 6 inches The reaction solution was poured into ice water, acidified with dilute hydrochloric acid and slowly adjusted PH2-3, to precipitate a solid. Filtration, 70 ° C drying crude, the resulting crude product ko ko acid ester 300ml heating beating again. Filtered, 70 ° C dry. Recrystallization from DMF telmisartan of formula III as shown in 25. Ig, yield: 50%..........................
PATENT http://www.google.com/patents/WO2010146187A2?cl=en
For example, WO 2004/087676 describes the hydrolysis of a compound with the chemical name 4 '-((1,7'- dimethyl-2 ' -propyl-lH, 3 'H-2, 5 ' -bibenzo [d] imidazol-3 ' -yl) - methyl) biphenyl-2-carbonitrile and having formula 2


Example 3: Isolation of telmisartan Into a reaction vessel 7.5g (15 mmol) of cyanotelmisartan, 30 ml of propylene glycol, 0.8 ml of water and 3g (45 mmol) of 85% KOH were added. The reaction mixture was heated to around 1600C to 170 0C and stirred at this temperature for 24 h. The reaction mixture was cooled below 800C and 75 ml of water were added. Then, pH value of the mixture was adjusted to 4.8 (by addition of 6M HCl) and then 150 ml of dichloromethane were added. The mixture was stirred for approximately 5 min and then the phases were separated. The water phase is reextracted by 50 ml of dichloromethane. Collected organic phases were washed with water (2χ50ml) and then treated with activated charcoal (2 g) . After that the organic phase was evaporated to an oily residue (9.8g) . 100 ml of acetone were added. The mix- ture was stirred at room temperature for at least 6 hours. The precipitated product was separated and washed with fresh acetone and dried at 65°C under reduced pressure for 3 hours. Yield: 6.8g (88%) Area % HPLC: Telmisartan 99.60%
.........................
PATENT http://www.google.com/patents/CN1548421A?cl=en
Specific embodiments 14 'Example - [(1,4'-dimethyl-2'-propyl [2,6'- two-1H - benzoimidazol] 1'-yl) methyl] - [1, 1'-biphenyl] -2-carboxylic acid sodium salt in 250ml reaction flask, telmisartan 10g (0.0195mol), NaOH0.75g (0.0189mol) and water 100ml, stirred for 1 hour (30 ℃), filtered insoluble materials are removed and concentrated to a small volume, plus ethanol 30ml, concentrated, washed with 30ml of n-hexane, decanted, plus ethanol 30ml, concentrated, and then repeat again, and concentrated to dryness to obtain telmisartan sodium salt 9.9g yield 95.2%. Melting point: 223-225 ℃. Elemental analysis: C33H29N4O2Na · H2O Calcd: C71.48 H5.10 N10.11 Found: C71.42 H5.08 N10.22
Example 24 '- [(1,4'-dimethyl-2'-n propyl [2,6'- two-1H - benzoimidazol] 1'-yl) methyl] - [1,1'-biphenyl] -2-carboxylic acid potassium salt in 250ml reaction flask, Telmisartan 10g (0.0195mol), KOH1.06g (0.0188mol) and water 100ml, stirred for 1 hour (30 ℃), filtered to remove insolubles, and concentrated to a small volume, ethanol 30ml, concentrated, hexane 30ml washed, decanted, plus ethanol 30ml, concentrated, and then repeat again, and concentrated to dryness to obtain telmisartan potassium 10.6g, yield 95.6%. Melting point: 203-205 ℃. Elemental analysis: C33H29N4O2K · H2O Calcd: C69.04 H5.40 N9.76 Found: C69.01 H5.28 N9.88
Example 3 starting material and the mixed powder was sieved excipients, 5% polyethylene pyrrolidone was granulated and dried. After dried particles were sieved magnesium stearate was added mixed tabletted. mg / tablet of telmisartan sodium salt 20 Lactose 170 Sodium carboxymethyl starch 10 mg Magnesium stearate 8 meglumine 25% polyvinyl pyrrolidone solution q.s.
Example 4 A mixed powder of raw materials and auxiliary materials sieved, added 5 % solution of polyvinylpyrrolidone is granulated and dried. After dried particles were sieved magnesium stearate was added mixed tabletted. mg / tablet telmisartan sodium Lactose 200 40 140 DCP sodium carboxymethyl starch 16 mg Magnesium stearate 45% povidone solution appropriate amount of
Example 5 of this product, according to the dissolution assay (Chinese Pharmacopoeia 2000 edition Appendix II XC second method), phosphate buffer 900ml solvent, the speed of 75 revolutions per minute, operate according to the law, after 30 minutes, take the solution as spectrophotometry (Chinese Pharmacopoeia 2000 edition of the test solution, according to the spectrophotometric two Appendix IVA), absorbance was measured at 295nm wavelength. Another reference standard stock solution 10ml precise amount of determination under set 100ml flask, diluted with phosphate buffer to the mark, then the precise amount of 5ml, set 10ml volumetric flask, dilute to the mark with phosphate buffer , shake, the same method absorption, calculated for each piece of the dissolution of the limits of 80% scalar, should be specified. Dissolution test results in Table. Table dissolution test results Dissolution (%) telmisartan sodium 97.29 99.65 102.55 95.83 101.10 98.92 99.20 ± 2.45
.......................
PATENT http://www.google.com/patents/CN1412183A?cl=en
Example 5 4 '- [(1,4'-dimethyl-2'-propyl [2,6'- two -1H- benzimidazol] -1'-yl) methyl] - [1,1' - biphenyl] -2-carboxylic acid (III) IV (24.8g, 0.05mol) was added ethylene glycol (100ml) and water (150ml) (or other previously described a mixed solvent), sodium ethoxide (or as previously said other alcohols sodium) (13.6g, 0.2mol), was refluxed for 10 hours. After no starting material by TLC was cooled to room temperature, hydrochloric acid was added dropwise (1/1) to pH 5-6, the precipitated solid was filtered, washed with water to give III.
........................
PATENT http://www.google.com/patents/CN101550107B?cl=en Example 3 [0047] 1) Preparation of telmisartan crude methyl esterCompound II into 50g in 500mL reaction flask, 200mL of methyl isobutyl ketone (MIBK), 25 ° C _30 ° C with stirring until dissolved, was added dropwise 35mL of triethylamine was added 55. Og After the completion of the compound III, 5 (T60 ° C or so for about 4_5 hours, TLC monitoring completion of the reaction, filtered and the filter cake washed with a small amount of MIBK, and then washed with water, dried to give 70. 3g of crude product. 81% yield, purity of about 98%. (TLC test conditions: ethyl acetate: methanol = 8: 1) 2) preparation of high purity methyl telmisartan IOOOmL reaction flask, the input step to give the crude methyl ester telmisartan, add 500mL of isopropanol was heated to dissolve, 2gX 2 activated bleaching filtrate was heated to about 90 ° C, added dropwise with stirring 150mL 7jC insulation 0. 5~Ih, cooled slowly to room temperature with stirring. Filtered, and the filter cake washed sequentially with MIBK and water washing, and drying, the yield of about 82%, HPLC purity 99.5%, the single impurities less than 0.1%. 3)
Preparation of telmisartan with high purity [0053] A reaction flask was put in a 500mL high purity 15g telmisartan ester, 3. Og sodium, 200mL of isopropanol, water, 80ml, was heated to reflux for 5 ~ 7 h, TLC monitoring of the reaction was complete, the distillation Isopropanol was removed, and water was added to completely dissolve the solid 40ml, 0. 5g of activated carbon bleaching, the filtrate was added 50ml of water, heated to 80 ° C, lmol / L of acetic acid to adjust the pH to 5. (Γ5. 5, filtered, and the filter cake dried to give 13. 14g of solid, yield 90%, HPLC purity 99.7%, the single impurities less than 0.1%. (TLC test conditions: ethyl acetate: methanol = 8: 1)
.............................
http://www.google.com/patents/CN101172968B?cl=en
Example 1 [0023] 1, 100gPPA, 21. 8g (0. Lmol) 2_ n-propyl _4_ _6_ carboxyl methyl benzimidazole and 21. 5gN- methyl-o-phenylenediamine added to the reaction flask in under N2 protection feeding, heated to IO (TC _1601 :, reaction 8-20 hours, down 70-80.C 200ml water was added and the reaction with hydrochloric acid to adjust ffl = 1~2, put charcoal 5_8%,, 8 (TC about 5 to 10 minutes, filtered, and the reaction repeated, the adjustment ra 12-14 with NaOH, for several hours, and filtered to give the crude intermediate 2-n-propyl -4-methyl-6- (benzimidazol-2-yl-methyl ) benzimidazole sodium salt. [0024] 2, the product of the previous step, 2-n-propyl -4-methyl--6_ (methyl benzimidazol-_2_ yl) benzimidazole sodium salt crude product was dissolved into 200 ml of ethanol , and dissolved by heating, cooling to room temperature, 400 ml 1N NaOH, to precipitate the compound 2-n-propyl -4-methyl-6- (methyl benzimidazol-2-yl) benzimidazole .50-8 ( TC dried in vacuo. [0025] 3, product of the previous step -4-methyl-2-n-propyl -6_ (methyl benzimidazol-_2_ yl) benzimidazole into 200 ml of dimethyl sulfoxide was stirred was added at room temperature and 4-bromomethyl - biphenyl-2-carboxylic acid methyl ester 33.55 g, was stirred for 14 hours, extracted with dichloromethane (200, 100, 100), and evaporated to dryness under reduced pressure, 300 ml of methanol and 10% potassium hydroxide (240 ml, 0. 6mo1) mixture was refluxed for 6 hours, cooled, washed with 80 ml of methylene chloride, adjusted with glacial acetic acid ffl = 6, a lot of white floc precipitated precipitate was filtered and dried to give a white Tilmicosin 49.6 g of crude product, the crude product was added 100 ml of chloroform was heated to reflux, activated carbon decolorization, crystallization, filtration, 8 (TC dried in vacuo to give a white pure telmisartan (HLPC> 99. 0%) 41 克, purification yield 82%. mp 261~263.C, H-NMR (d6-DMS0) S 1. 05t, 3H), 1. 83 (m, 2H), 2. 71 (s, 3H), 2. 94 (t, 2H), 3. 81 (s, 3H), 5. 57 (s, 2H), 7. 16-7. 83 (m, 14H) • C33H33N402 [0026]
Example 2 Preparation of telmisartan 1, 100gPPA, 21. 8g (0. 1) 2_ [4-methyl-n-propyl-benzimidazole and _6_ 21. 5gN- carboxy-o-phenylenediamine added to the reaction flask in N2 Under the protection of feeding, heated to 100 ° C _160 ° C, the reaction for 8-20 hours, down 70-80. C, the reaction was added 200ml of water, adjusted with hydrochloric acid ffl = 1~2, into charcoal 5_8%, about 8 (TC 5_10 minutes filtered again reacted with K0H ra adjusted to 12-14 for several hours and filtered to give Intermediate crude 2-n-propyl -4-methyl-6- (benzimidazol-2-yl-methyl) benzimidazole potassium salt.
2, the product of the previous step, 2-n-propyl -4-methyl--6_ (methyl benzimidazol-_2_ yl) benzimidazole potassium salt of the crude product into 200 ml of ethanol, and dissolved by heating, cooling to room temperature was added 400 ml 1N K0H, a precipitated compound is 2-n-propyl -4-methyl-6- (benzimidazol-2-yl-methyl) benzimidazole potassium salt. 50-8 (TC dried in vacuo.
[0029] 3, 2-n-propyl prepared in the previous step -4-methyl-6- (benzimidazol-2-yl-methyl) benzimidazole potassium salt and 27.2 g of 4-bromomethyl-2-cyanobiphenyl, 10.4 g of triethylamine and DMF (DMA, dichloromethane, dichloroethane) were mixed and reacted for 5-10 hours at 35-40 °, TLC detection After no starting material the reaction mixture was poured into 600 g of ice water, extracted with ethyl acetate (300ml * 3), the combined organic phases were washed with water (300ml * 2), dried and desolvation, and then petroleum ether was added and stirred until a solid precipitated was The crude product was 45.6 g.
4, the upper step of the solid 45.6 grams, was added 200ml of ethylene glycol, 150ml water, 12 g of sodium hydroxide, the reaction was refluxed for 10 hours, TLC detected no starting material and then cooled to room temperature, acidified with hydrochloric ra is 5 to 6, there is solid precipitation, filtration, washing, telmisartan was crude, DMF and recrystallized to give 44.5 g of telmisartan pure product (HLPC> 99. 0%) mp261~263 ° C. Force -NMR (de-DMS0) S 1. 05t, 3H), 1. 83 (m, 2H), 2. 71 (s, 3H), 2. 94 (t, 2H), 3. 81 (s, 3H ), 5. 57 (s, 2H), 7. 16-7. 83 (m, 14H) • C33H33N402 [0031] ............................
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| CN100460396C | Mar 8, 2007 | Feb 11, 2009 | 杭州盛美医药科技开发有限公司 | Intermediate of telmisartan, preparation and use thereof |
| CN100506799C | Apr 19, 2007 | Jul 1, 2009 | 北京理工大学 | [(2-n-propyl-4-methyl-1H-Benzimidazole)6-radical] carboxamide-1-radical] methylbiphenyl compound, synthesizing method and usage |
| CN100548293C | Aug 8, 2003 | Oct 14, 2009 | 贝塞斯达药物股份有限公司 | Novel PPAR ligands that do not cause fluid retention, edema or congestive heart failure |
| CN101550107B | Apr 2, 2009 | Jan 12, 2011 | 宁波九胜创新医药科技有限公司 | Method for preparing telmisartan |
| CN101743228B | Jul 3, 2008 | Jan 29, 2014 | 新梅斯托克尔卡托瓦纳兹德拉韦尔公司 | Process for preparing telmisartan |
| CN101891735B | Nov 25, 2009 | Jul 18, 2012 | 北京理工大学 | Biphenyl sulfafurazole compound, synthesis method and application thereof |
| CN102093297B | Jan 28, 2011 | Aug 1, 2012 | 海南美兰史克制药有限公司 | Telmisartan compound and new preparation method thereof |
| EP1805146A2 * | Oct 18, 2005 | Jul 11, 2007 | Dr. Reddy's Laboratories Ltd. | Process for preparing telmisartan |
| EP2123648A1 | May 20, 2008 | Nov 25, 2009 | Chemo Ibérica, S.A. | A process for the preparation of Telmisartan. |
| US7501448 | Oct 13, 2005 | Mar 10, 2009 | Teva Pharmaceutical Industries, Ltd. | high yields, low cost process; easy solvent extraction; nontoxic, safe, environmentally friendly, low boiling point organic solvents; 1,7'-dimethyl-2'-propyl-1H,3'H-[2,5']bibenzoimidazolyl is reacted with 4'bromomethyl-biphenyl-2-carboxylic acid alkyl ester; industrial scale; hydrolysis |
| US8691999 * | May 10, 2005 | Apr 8, 2014 | Cipla Limited | Process for the preparation of telmisartan |
| WO2006044648A1* | Oct 13, 2005 | Apr 27, 2006 | Teva Pharma | Process for preparing telmisartan |
| WO2007010558A1* | Jul 19, 2006 | Jan 25, 2007 | Satyanarayana Chava | A process for the preparation of telmisartan |
| WO2009123483A1 | Mar 30, 2009 | Oct 8, 2009 | Zaklady Farmaceutyczne Polpharma Sa | Process for preparation of telmisartan |
| WO2012028925A2 | Aug 29, 2011 | Mar 8, 2012 | Ogene Systems (I) Pvt Ltd | An improved process for the preparation of telmisartan |
| CN1412183A | Oct 15, 2001 | Apr 23, 2003 | 中国科学院上海药物研究所 | New preparation method of timixatan |
| CN1620437A | Jan 15, 2003 | May 25, 2005 | 贝林格尔英格海姆法玛两合公司 | Method for the production and purification of 1, 7'-dimethyl-2'-propyl-2, 5'-bi-1h-benzimidazole |
| CN1768044A | Mar 26, 2004 | May 3, 2006 | 贝林格尔·英格海姆国际有限公司 | Process for manufacture of telmisartan |
| WO2005108375A1 | May 10, 2005 | Nov 17, 2005 | Cipla Ltd | Process for the preparation of telmisartan |
| CN1344712A | Jul 30, 2001 | Apr 17, 2002 | 中国科学院上海药物研究所 | Synthesis path of Timisatem | |
| CN1412183A | Oct 15, 2001 | Apr 23, 2003 | 中国科学院上海药物研究所 | New preparation method of timixatan | |
| US2006/0094883 | Title not available | ||||
| WO03/059890A1 | Title not available | ||||
| WO2005/108375A1 | Title not available |
| US7501448 | Oct 13, 2005 | Mar 10, 2009 | Teva Pharmaceutical Industries, Ltd. | high yields, low cost process; easy solvent extraction; nontoxic, safe, environmentally friendly, low boiling point organic solvents; 1,7'-dimethyl-2'-propyl-1H,3'H-[2,5']bibenzoimidazolyl is reacted with 4'bromomethyl-biphenyl-2-carboxylic acid alkyl ester; industrial scale; hydrolysis |
| WO2007147889A2* | Jun 22, 2007 | Dec 27, 2007 | Krka Tovarna Zdravil D D Novo | Preparation of telmisartan salts |
| WO2010146187A2 | Jun 21, 2010 | Dec 23, 2010 | Krka, Tovarna Zdravil, D.D., Novo Mesto | Process for the preparation of telmisartan |
| WO2012055941A1 | Oct 26, 2011 | May 3, 2012 | Krka,Tovarna Zdravil, D. D., Novo Mesto | Multilayer pharmaceutical composition comprising telmisartan and amlodipine |
| WO2003007876A2 | Jun 25, 2002 | Jan 30, 2003 | Sumner H Burstein | N-fatty acid-amino acid conjugates and therapeutic uses |
| WO2003059327A1 | Jan 16, 2002 | Jul 24, 2003 | Boehringer Ingelheim Pharma | Bilayer pharmaceutical tablet comprising telmisartan and a diuretic and preparation thereof |
| WO2004028505A1 | Sep 18, 2003 | Apr 8, 2004 | Boehringer Ingelheim Int | Solid pharmaceutical formulations comprising telmisartan |
| WO2004087676A1 | Mar 26, 2004 | Oct 14, 2004 | Boehringer Ingelheim Int | Method for the production of telmisartan |
| WO2004096215A1 | Apr 27, 2004 | Nov 11, 2004 | Boehringer Ingelheim Int | Pharmaceutical formulation of the sodium salt of telmisartan |
| WO2005108375A1* | May 10, 2005 | Nov 17, 2005 | Cipla Ltd | Process for the preparation of telmisartan |
| WO2006044754A2 | Oct 18, 2005 | Apr 27, 2006 | Muthulingam Arunagiri | Process for preparing telmisartan |
| WO2006050509A2 | Nov 3, 2005 | May 11, 2006 | Teva Pharma | Amorphous and polymorphic forms of telmisartan sodium |
| WO2006050921A2 | Nov 9, 2005 | May 18, 2006 | Lek Pharmaceuticals | Preparation of telmisartan salts with improved solubility |
| WO2006063737A1 | Dec 9, 2005 | Jun 22, 2006 | Boehringer Ingelheim Int | Combination therapy comprising telmisartan and hydrochlorothiazide |
| WO2006136916A2 | Jun 20, 2006 | Dec 28, 2006 | Glenmark Pharmaceuticals Ltd | Substantially pure micronized particles of telmisartan and pharmaceutical compositions containing same |
| WO2007010559A2 | Jul 19, 2006 | Jan 25, 2007 | Panacea Biotec Ltd | Novel pharmaceutical modified release dosage form cyclooxygenase enzyme inhibitor |
| WO2007060170A2 | Nov 22, 2006 | May 31, 2007 | Boehringer Ingelheim Int | Bilayer tablet comprising telmisartan and diuretic |
| WO2007144175A2 | Jun 14, 2007 | Dec 21, 2007 | Lek Pharmaceuticals | Pharmaceutical composition comprising hydrochlorothiazide and telmisartan |
| WO2007147889A2 | Jun 22, 2007 | Dec 27, 2007 | Krka Tovarna Zdravil D D Novo | Preparation of telmisartan salts |
| WO2009004064A1 | Jul 3, 2008 | Jan 8, 2009 | Krka Tovarna Zdravil D D Novo | Process for preparing telmisartan |
| CN1412183A | Oct 15, 2001 | Apr 23, 2003 | 中国科学院上海药物研究所 | New preparation method of timixatan |
| CN1548421A | May 22, 2003 | Nov 24, 2004 | 上海医药工业研究院 | Tilmisartan salt and its prepn |
| EP0502314A1 | Jan 31, 1992 | Sep 9, 1992 | Dr. Karl Thomae GmbH | Benzimidazol, medicaments containing them and process for their preparation |
| EP1144386A1 | Jan 7, 2000 | Oct 17, 2001 | Boehringer Ingelheim Pharma KG | Telmisartan polymorphs, methods for producing same and their use in the preparation of a medicament |
| EP1719766A2 | Apr 18, 2006 | Nov 8, 2006 | Dipharma S.p.A. | A process for the preparation of telmisartan |
| US20060264491 | Jun 8, 2006 | Nov 23, 2006 | Chemagis Ltd. | Telmisartan production process |
| CN101172968B | Nov 1, 2006 | May 12, 2010 | 浙江天宇药业有限公司 | 2-propyl-4 methyl-6-(tolimidazole-2group) benzoglioxaline salt and method for producing the same |
| CN101743228B | Jul 3, 2008 | Jan 29, 2014 | 新梅斯托克尔卡托瓦纳兹德拉韦尔公司 | Process for preparing telmisartan |
| CN102015690B | Mar 19, 2009 | Apr 30, 2014 | 力奇制药公司 | Catalyzed carbonylation in the synthesis of angiotensin II antagonists |
| CN102036937B | Mar 19, 2009 | Jun 4, 2014 | 力奇制药公司 | 2'-halobiphenyl-4-yl intermediates in the synthesis of angiotensin ii antagonists |
| WO2014067237A1* | Oct 31, 2013 | May 8, 2014 | Topharman Shanghai Co., Ltd. | Telmisartan preparation method and intermediate thereof |
| WO2010018441A2* | Aug 10, 2009 | Feb 18, 2010 | Cadila Pharmaceuticals Ltd. | An improved process for the preparation of substantially pure telmisartan |
| WO2010146187A2* | Jun 21, 2010 | Dec 23, 2010 | Krka, Tovarna Zdravil, D.D., Novo Mesto | Process for the preparation of telmisartan |
| WO2011077444A1* | May 28, 2010 | Jun 30, 2011 | Inogent Laboratories Private Limited | A new process for the preparation of pure telmisartan |
| WO2012028925A2* | Aug 29, 2011 | Mar 8, 2012 | Ogene Systems (I) Pvt Ltd | An improved process for the preparation of telmisartan |
| CN1768044A * | Mar 26, 2004 | May 3, 2006 | 贝林格尔·英格海姆国际有限公司 | Process for manufacture of telmisartan |
| CN102731407A * | Jul 4, 2012 | Oct 17, 2012 | 宁波九胜创新医药科技有限公司 | Method for preparing telmisartan |
| EP0627433A1 * | Dec 7, 1993 | Dec 7, 1994 | Eisai Co., Ltd. | Process for producing imidazopyridine derivative and intermediate |
| EP2123648A1 * | May 20, 2008 | Nov 25, 2009 | Chemo Ibérica, S.A. | A process for the preparation of Telmisartan. |
| EP2305650A1 * | Sep 21, 2009 | Apr 6, 2011 | Chemo Ibérica, S.A. | Novel process for the preparation of telmisartan |
| KR20090000113A * | Title not available | |||
| US20040236113 | Mar 17, 2004 | Nov 25, 2004 | Boehringer Ingelheim International Gmbh | Process for manufacture of telmisartan |
| US20130137878 | Jan 25, 2013 | May 30, 2013 | Boehringer Ingelheim International Gmbh | Process for manufacture of telmisartan |
I notice this topic and I know it is complete, but I read it carefully since it is a really essential issue. However, I go to Best Essay Writer, and this service solves all of my difficulties. So I go to this service and look at the material on this issue.
ReplyDeleteYes, You done really nice work in this article post. I apricate that. I hope in future you share more good updates. Now it's time to avail Baby Liquid Soap for more information.
ReplyDeleteIt is a common medicine used by everyone. I must appreciate the effort you have put in this article. Please keep me updated in the post. Excellent job. Keep the hard work going. Nice work. Now it's time to avail Best skincare cream for more information.
ReplyDeleteMATLAB Assignment Help offers professional assistance to students struggling with complex mathematical modelling, simulations, coding, data analysis, and algorithm development in MATLAB. Expert programmers and engineers provide accurate, well-documented solutions tailored to university requirements, whether it’s signal processing, image processing, control systems, numerical methods, or machine learning tasks. The service ensures error-free code, clear explanations, and timely delivery to help you understand concepts while scoring better grades. With 24/7 support and personalised guidance, MATLAB Assignment Help makes technical learning easier and stress-free.
ReplyDelete