
Pasireotide, Signifor; SOM 320; HY-16381; 396091-73-9
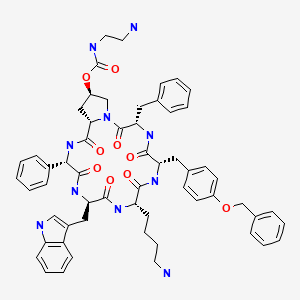
Cyclo[4(R)-[N-(2-aminoethyl)carbamoyloxy]-L-prolyl-L-phenyl-glycyl-D-tryptophyl-L-lysyl-(4-O-benzyl)-L-tyrosyl-L-phenylalanyl]bis(L-aspartic acid)
Regulators in the USA has approved a long-acting release of Novartis’ Signifor as a treatment for acromegaly.
The Food and Drug Administration has approved Signifor LAR (pasireotide) for the treatment of patients with acromegaly who have had an inadequate response to surgery or for whom the latter is not an option. The thumbs-up comes a month after the European Medicines Agency approved the drug, a next-generation somatostatin analogue administered intramuscularly once-monthly.
Read more at: http://www.pharmatimes.com/Article/14-12-16/FDA_gives_green_light_to_Novartis_acromegaly_drug.aspx#ixzz3M8Ibn14Q
Pasireotide (SOM230, trade name Signifor[1]) is an orphan drug approved in the U.S. and Europe for the treatment of Cushing’s disease in patients who fail or are ineligible for surgical therapy.[2][3] It was developed by Novartis. Pasireotide is a somatostatinanalog which has a 40-fold increased affinity tosomatostatin receptor 5 than other somatostatin analogs.
The drug showed therapeutical potential in a recent study (PASPORT-CUSHINGS B2305) where 162 patients were treated with either 2x 600 µg or2x 900 µg pasireotide s.c. daily.[4] The effectiveness of the treatment was checked by the UFC-value (urinary free cortisol) after six months of treatment. The mean reduction of UFC after six months was 47.9%, which also lead to amelioration of clinical symptoms such as blood pressure, cholesterol value, and weight loss.[5]
At present, it is in phase III clinical trials at Novartis for the treatment of carcinoid tumors and symptoms that are not adequately controlled by somatostatin analogues (Sandostatin). Phase II clinical development is also under way at the company for the treatment of gastric dumping syndrome, metastatic carcinoid tumors, meningioma and pituitary adenoma and for the treatment of hepatocellular carcinoma in combination with everolimus. Early clinical trials are also ongoing for the treatment of patients with metastatic melanoma or Merkel cell carcinoma. A phase I clinical trial for the treatment of alcoholic cirrhosis has been completed. The company intends to file for approval in 2007 for these indications. Novartis and Thomas Jefferson University are conducting phase II clinical trials for the treatment of prostate cancer, alone or in combination with everolimus. The Mayo Clinic is conducting phase II clinical trials for the treatment of polycystic liver disease. Phase III clinical trials had been ongoing for the reduction of post-pancreatectomy fistula, leak, and abscess; however, in 2010 these trials were suspended. In 2004, orphan drug designation was assigned in the E.U. for the treatment of functional gastroenteropancreatic endocrine tumors. In 2009, orphan drug designation was received in the U.S. and the E.U. for the treatment of Cushing’s disease and acromegaly. The designation for the treatment of Cushing’s disease was assigned in Australia in 2011 and in Japan in 2012. In 2013, orphan drug designation was assigned in Australia for the treatment of acromegaly.

SIGNIFOR (pasireotide diaspartate) injection is prepared as a sterile solution of pasireotide diaspartate in a tartaric acid buffer for administration by subcutaneous injection. SIGNIFOR is a somatostatin analog. Pasireotide diaspartate, chemically known as (2-Aminoethyl) carbamic acid (2R,5S,8S,11S,14R,17S,19aS)-11-(4-aminobutyl)-5-benzyl-8-(4-benzyloxybenzyl)-14-(1H-indol-3ylmethyl)-4,7,10,13,16,19-hexaoxo-17-phenyloctadecahydro-3a,6,9,12,15,18hexaazacyclopentacyclooctadecen-2-yl ester, di[(S)-2-aminosuccinic acid] salt, is a cyclohexapeptide with pharmacologic properties mimicking those of the natural hormone somatostatin.
The molecular formula of pasireotide diaspartate is C58H66N10O9 • 2C4H7NO4and the molecular weight is 1313.41. The structural formula is:
 SIGNIFOR is supplied as a sterile solution in a single-dose, 1 mL colorless glass ampule containing pasireotide in 0.3 mg/mL, 0.6 mg/mL, or 0.9 mg/mL strengths for subcutaneous injection.
SIGNIFOR is supplied as a sterile solution in a single-dose, 1 mL colorless glass ampule containing pasireotide in 0.3 mg/mL, 0.6 mg/mL, or 0.9 mg/mL strengths for subcutaneous injection.
Each glass ampule contains:
| 0.3 MG | 0.6 MG | 0.9 MG | |
| Pasireotide diaspartate | 0.3762* | 0.7524* | 1.1286* |
| Mannitol | 49.5 | 49.5 | 49.5 |
| Tartaric acid | 1.501 | 1.501 | 1.501 |
| Sodium hydroxide | ad pH 4.2 | ad pH 4.2 | ad pH 4.2 |
| Water for injection | ad 1ml | ad 1ml | ad 1ml |
| * corresponds to 0.3/0.6/0.9 mg pasireotide base Note: Each ampule contains an overfill of 0.1ml to allow accurate administration of 1 ml from the ampule. | |||
 | |
| SYSTEMATIC (IUPAC) NAME | |
|---|---|
| [(3S,6S,9S,12R,15S,18S,20R)-9-(4-aminobutyl)-3-benzyl-12-(1H-indol-3-ylmethyl)-2,5,8,11,14,17-hexaoxo-15-phenyl-6-[(4-phenylmethoxyphenyl)methyl]-1,4,7,10,13,16-hexazabicyclo[16.3.0]henicosan-20-yl] N-(2-aminoethyl)carbamate | |
| CLINICAL DATA | |
| TRADE NAMES | Signifor |
| LICENCE DATA | EMA:Link |
| LEGAL STATUS |
|
| ROUTES | Subcutaneous |
| IDENTIFIERS | |
| CAS NUMBER | 396091-73-9 |
| ATC CODE | H01CB05 |
| PUBCHEM | CID 9941444 |
| UNII | 98H1T17066 |
| SYNONYMS | SOM230 |
| CHEMICAL DATA | |
| FORMULA | C58H66N10O9 |
| MOL. MASS | 1107.26 g/mol |
Pasireotide is a multiligand somatostatin analogue with high binding affinity to somatostatin receptors sst1, sst2, sst3 and sst5. Novartis Oncology, a division of Novartis, filed for approval in the E.U. for the treatment of Cushing’s syndrome in 2010. A positive opinion was granted in 2011 and final approval was obtained in 2012. The E.U.’s first launch took place in Germany in June 2012. Also in 2011, Novartis filed an NDA in the U.S. seeking approval of the compound for the treatment of Cushing’s syndrome; however, the application was withdrawn the same year due to an issue related to chemistry, manufacturing and controls. In November 2012, the product was recommended for approval in the U.S. for Cushing’s syndrome. In December 2012, final FDA approval was granted. Phase III clinical trials are ongoing in Japan for this indication. In 2014, the product was approved in the E.U and the U.S. for the treatment of adult patients with acromegaly for whom surgery is not an option or has not been curative and who are inadequately controlled on treatment with a first-generation somatostatin analogue (SSA).
- http://www.google.com/patents/EP2310042B1?cl=en
- The present invention relates to a new use of Somatostatin (SRIF) peptidomimetics (also referred to as Somatostatin- or SRIF-analogs).
- Somatostatin is a tetradecapeptide having the structure

- The somatostatin class is a known class of small peptides comprising the naturally occurring somatostatin-14 and analogues having somatostatin related activity, e.g. as disclosed by A.S. Dutta in Small Peptides, Vol.19, Elsevier (1993). By “somatostatin analog” as used herein is meant any straight-chain or cyclic polypeptide having a structure based on that of the naturally occurring somatostatin-14 wherein one or more amino acid units have been omitted and/or replaced by one or more other amino radical(s) and/or wherein one or more functional groups have been replaced by one or more other functional groups and/or one or more groups have been replaced by one or several other isosteric groups. In general, the term covers all modified derivatives of the native somatostatin-14 which exhibit a somatostatin related activity, e.g. they bind to at least one of the five somatostatin receptor (SSTR), preferably in the nMolar range.
- Natural somatostatin binds and activates all 5 somatostatin receptors (SSTR1-5) with nmol efficacy and thus causes its multiple physiological effects.
- Synthetically available somatostatin analogs differ in their binding affinity to the different somatostatin receptor subtypes and often bind selectively to one or few subtypes with significantly higher affinity.
- Somatostatin analogs of particular interest according to the present invention have a high binding affinity to human SSTR1,2,3,5 and have been described e.g. in WO 97/01579 , the contents of which being incorporated herein by reference. Said somatostatin analogs comprise the amino acid sequence of formula I-(D/L)Trp-Lys-X1 -X2 - Iwherein X1 is a radical of formula (a) or (b)
 wherein R1 is optionally substituted phenyl, wherein the substituent may be halogen, methyl, ethyl, methoxy or ethoxy,
wherein R1 is optionally substituted phenyl, wherein the substituent may be halogen, methyl, ethyl, methoxy or ethoxy,
R2 is -Z1-CH2-R1, -CH2-CO-O-CH2-R1, wherein Z1 is O or S, and
wherein Z1 is O or S, and
X2 is an α-amino acid having an aromatic residue on the Cα side chain, or an amino acid unit selected from Dab, Dpr, Dpm, His,(Bzl)HyPro, thienyl-Ala, cyclohexyl-Ala and t-butyl-Ala, the residue Lys of said sequence corresponding to the residue Lys9 of the native somatostatin-14. - Somatostatin analogs of particular interest which have a high binding affinity to human SSTR1,2,3,5 have also been described e.g. inWO02/10192. Said somatostatin analogs comprise the compound of formula
 also called cyclo[{4-(NH2-C2H4-NH-CO-O-)Pro}-Phg-DTrp-Lys-Tyr(4-Bzl)-Phe] or pasireotide, as well as diastereoisomers and mixtures thereof, in free form, in salt or complex form or in protected form. Phg means -HN-CH(C6H5)-CO- and Bzl means benzyl.
also called cyclo[{4-(NH2-C2H4-NH-CO-O-)Pro}-Phg-DTrp-Lys-Tyr(4-Bzl)-Phe] or pasireotide, as well as diastereoisomers and mixtures thereof, in free form, in salt or complex form or in protected form. Phg means -HN-CH(C6H5)-CO- and Bzl means benzyl.
…………………
Example 1 : Cyclo[{4-(NH2-C2H4-NH-CO-O-
a) Synthesis of Fmoc-Pro(4-OCO-NH-CH2-CH2-NH-Boc)-OH
L-hydroxyproline methylester hydrochloride is reacted with Fmoc-OSu in aqueous 1.0 N sodium carbonate/THF at room temperature. After completion of the reaction, Fmoc-Pro(4- OH)-OMe is isolated by precipitation. Fmoc-Pro(4-OH)-OMe is then added dropwise into a solution of trisphosgene (0.6 eq.) in THF to give a chlorocarbonate intermediate. After 1 h dimethylaminopyridine (1.0 eq.) and N-Boc-diaminoethane (6.0 eq.) are added and the reaction is stirred at room temperature. After completion of the reaction, the solvent is removed in vacuo and the resulting Fmoc-Pro(4-OCO-NH-CH2-CH2-NH-Boc)-OMe is extracted from a two phase system of ethyl acetate/0.1 M HCI to give crude product (MH+ = 554) which is purified by crystallization from ethyl acetate. The methyl ester is then cleaved to the free acid by treatment with 1 N NaOH in dioxane/water and the product Fmoc-Pro(4-OCO-NH-CH2-CH2-NH-Boc)-OH is purified on silica gel, [(M+Na)]+= 562).
b) H-Phe-Pro(4-OCO-NH-CH2-CH2-NH-Boc)-Phg-DTrp(Boc)-Lys(Boc)-Tyr(Bzl)-OH Commercially available Fmoc-Tyr(Bzl)-O-CH2-Ph(3-OCH3)-O-CH2-Polystyrene resin (SASRIN-resin, 2.4 mM) is used as starting material and carried through a standard protocol consisting of repetitive cycles of Nα-deprotection (Piperidine/DMF, 2:8), repeated washings with DMF and coupling (DIPCI: 4.8 mM/HOBT: 6mM, DMF). The following amino acid- derivatives are sequentially coupled: Fmoc-Lys(Boc)-OH, Fmoc-DTrp(Boc)-OH, Fmoc-Phg- OH, Fmoc-Pro(4-OCO-NH-CH2-CH2-NH-Boc)-OH, Fmoc-Phe-OH. Couplings (2 eq. amino acids) are continued or repeated until completion, i.e. until complete disappearance of residual amino groups which is monitored by a negative ‘Kaiser* Ninhydrin test. Before cleavage of the completely assembled protected linear peptide from its resin support the Nα-Fmoc protection from the last residue is removed.
c) H-Phe-Pro(4-OCO-NH-CH2-CH2-NH-Boc)-Phg-DTrp(Boc)-Lys(Boc)-Tyr(Bzl)-OH After washings with CH2CI2) the peptide-resin is transferred into a column or a stirred suction filter and the peptide fragment is cleaved and eluted with a short treatment with 2% TFA in CH2CI2 within 1 h. The eluate is immediately neutralized with a saturated NaHCO3 solution. The organic solution is separated and evaporated and the side chain protected precursor (MH+ = 1366) is cyclized without further purification.
d) cyclo[-Pro(4-OCO-NH-CH2-CH2-NH2)-Phg-DT -Lys-Tyr(Bzl)-Phe-], trifluoroacetate The above linear fragment is dissolved in DMF (4 mM), cooled to minus 5°C and treated with 2 eq. DIPEA then 1.5 eq. of DPPA and stirred until completion (ca. 20h) at 0-4°C. The solvent was almost completely removed in vacuo; the concentrate is diluted with ethyl acetate, washed with NaHCO3, water, dried and evaporated in vacuo.
For deprotection the residue is dissolved at 0°C in TFA H2O 95:5 (ca.50 mM) and stirred in the cold for 30 min. The product is then precipitated with ether containing ca. 10 eq. HCI, filtered, washed with ether and dried. In order to completely decompose remaining Indole-N carbaminic acid the product is dissolved in 5% AcOH and lyophilized after 15 h at ca. 5°C. Preparative RP-HPLC is carried out on a C-18 10 μm STAGROMA column (5-25 cm) using a gradient of 0.5% TFA to 0.5% TFA in 70% acetonitrile. Fractions containing the pure title compound are combined, diluted with water and lyophilized. The lyophilisate is dissolved in water followed by precipitation with 10% Na2CO3 in water. The solid free base is filtered of, washed with water and dried in vacuum at room temperature. The resulting white powder is directly used for the different salts.
Example 2: Cyclo[{4-(NH2-C2H4-NH-CO-O-)Pro}-Phg-DTrp-Lys-Tyr(4-Bzl)-Phe] in salt form a. Acetate
Conversion to the acetate salt form is carried out using an ion-exchange resin (e.g. AG 3- X4). MS (ESI): m/z 524.5 [M+2H]2+ [α]D 20= -42°, c=0.26 in AcOH 95%
b. Aspartate
Conversion to the mono- or di-aspartate is obtained by reacting 1 equivalent of the compound of Example 1 with 1 or 2 equivalent of aspartic acid in a mixture of acetonitrile/water 1 :3. The resulting mixture is frozen and lyophilized. The di-aspartate may also be obtained by dissolving the compound of Example 1 in water/acetonitrile 4:1, filtering, loading on a an ion-exchange resin, e.g. BioRad AG4X4 column, and eluting with water/acetonitrile 4:1. The eluate is concentrated, frozen and lyophilized. [ ]D 20= -47.5°, c= 2.5mg/ml in methanol
……………..
WO2013/174978 A1
………………………..
WO2013/131879 A1,
………………………..
WO2005/53732 A1,
……………………………
Journal of Medicinal Chemistry, 2003 , vol. 46, 12 pg. 2334 – 2344

A rational drug design approach, capitalizing on structure−activity relationships and involving transposition of functional groups from somatotropin release inhibitory factor (SRIF) into a reduced size cyclohexapeptide template, has led to the discovery of SOM230 (25), a novel, stable cyclohexapeptide somatostatin mimic that exhibits unique high-affinity binding to human somatostatin receptors (subtypes sst1−sst5). SOM230 has potent, long-lasting inhibitory effects on growth hormone and insulin-like growth factor-1 release and is a promising development candidate currently under evaluation in phase I clinical trials.
5.1.3.2. Cyclization, Deprotection, and Purification ofCyclo[(diaminoethylcarbamoyl)-HyPro-Phg-d-Trp-Lys-Tyr(Bzl)-Phe] (25).For cyclization, the above linear fragment was dissolved in DMF to a concentration of 4 mM, cooled to −5 °C, treated with 2 equiv of DIPEA and then 1.5 equiv of DPPA, and stirred at 0−4 °C until completion (ca. 20 h). The solvent was almost completely removed in vacuo. The concentrate was diluted with ethyl acetate, washed with NaHCO3 and water, dried, and evaporated in vacuo. The protected cyclized product was obtained in good yield.
For complete deprotection, the residue was dissolved at 0 °C in TFA/H2O, 95:5 (ca. 50 mM), and the mixture was stirred in the cold for 30 min. The product was then precipitated with ether containing ca. 10 equiv of HCl, filtered, washed with ether, and dried. To completely decompose the remaining indole-N carbaminic acid, the product was dissolved in 5% AcOH and lyophilized after 15 h at ca. 5 °C. Analytical RP-HPLC indicated a purity of 75% for the crude product.Preparative HPLC purification afforded 25: 3.1 g, 20% yield, purity 98%, RtI = 10.70, RtII = 10.20, RtIV = 3.90, HRMS 1047.51 (calcd 1047.5014).
Table 2. 1H and 13C NMR Assignments of SOM230, Using Numbering Scheme in NMR Assignment
| residue | group | δ 1H [ppm] | δ 13C [ppm] | residue | group | δ 1H [ppm] | δ 13C [ppm] |
| 1 | l-phenylglycine |
| 1 | NH | 9.73 | 1 | α-CH | 6.47 | 59.3 | |||
| 1 | 2/6-CH | 8.02 | 127.3 | 1 | CO | 169.6 | |||
| 1 | 3/5-CH | 7.41 | 129.1 | 1 | 1-C | 141.0 |
| 1 | 4-CH | 7.21 | 128.0 | |
| 2 | d-tryptophane |
| 2 | 1‘-NH | 12.20 | 2 | α-CH | 5.28 | 55.6 | |||
| 2 | NH | 10.34 | 2 | β-CH2 | 3.72 | 3.30 | 28.5 | ||
| 2 | 7-CH | 7.65 | 112.0 | 2 | CO | 173.9 | |||
| 2 | 4-CH | 7.43 | 119.2 | 2 | 8-C | 137.5 | |||
| 2 | 2-CH | 7.28 | 124.7 | 2 | 9-C | 128.3 | |||
| 2 | 6-CH | 7.23 | 121.6 | 2 | 3-C | 110.3 |
| 2 | 5-CH | 6.96 | 119.2 | |
| 3 | l-lysine |
| 3 | NH | 10.10 | 3 | δ-CH2 | 1.41 | 1.32 | 31.5 | ||
| 3 | α-CH | 4.62 | 55.2 | 3 | γ-CH2 | 0.89 | 23.5 | ||
| 3 | ε-CH2 | 2.80 | 41.0 | 3 | CO | 171.9 | |||
| 3 | β-CH2 | 1.87 | 1.32 | 31.6 | 3 | NH3+ | a |
| 4 | (4-O-benzyl)-l-tyrosine | |||
| 4 | NH | 7.99 | 4 | 7-CH2 | 4.92 | 69.9 | |||
| 4 | 2‘/6‘-CH | 7.46 | 128.0 | 4 | β-CH2 | 3.46 | 3.10 | 39.7 | |
| 4 | 3‘/5‘-CH | 7.37 | 128.9 | 4 | CO | 171.8 | |||
| 4 | 4‘-CH | 7.30 | 128.2 | 4 | 4-C | 157.9 | |||
| 4 | 2/6-CH | 7.21 | 131.5 | 4 | 1‘C | 137.9 | |||
| 4 | 3/5-CH | 6.85 | 114.7 | 4 | 1-C | 129.8 |
| 4 | α-CH | 5.23 | 53.1 | |
| 5 | l-phenylalanine |
| 5 | NH | 9.82 | 5 | α-CH | 4.42 | 53.9 | |||
| 5 | 2/6-CH | 7.38 | 130.0 | 5 | β-CH2 | 3.23 | 3.06 | 37.8 | |
| 5 | 3/5-CH | 7.27 | 129.3 | 5 | CO | 171.2 | |||
| 5 | 4-CH | 7.16 | 127.6 | 5 | 1-C | 136.3 |
| 6 | (γ-O-diaminoethylcarbamate)-l-hydroxyproline | ||||||
| 6 | 2-NH | 8.04 | 6 | 4-CH2 | 2.95 | 42.4 | |||
| 6 | γ-CH | 5.23 | 70.9 | 6 | β-CH2 | 2.63 | 1.25 | 37.0 | |
| 6 | α-CH | 4.22 | 60.6 | 6 | CO | 170.7 | |||
| 6 | δ-CH2 | 4.12 | 51.4 | 6 | 1-CO | 156.7 | |||
| 6 | 3-CH2 | 3.42 | 44.5 | 6 | 4-NH3+ | a |
| A | acetate |
| A | CH3 | 2.20 | 22.1 | A | CO | 174.3 |
a The NH3+ protons are part of the water peak at 5.82 ppm.
References
- Signifor® (pasireotide) Official Website for healthcare professionals outside the US http://www.signifor.com/
- “Novartis drug Signifor® approved in the EU as the first medication to treat patients with Cushing’s disease”. Retrieved 2012-07-08.
- Mancini et al. Therapeutics and Clinical Risk Management 2010;6:505-516
- Colao et al. Pasireotide (SOM230) provides clinical benefit in patients with Cushing’s disease: results from a large, 12-month, randomized-dose, double-blind, Phase III study, Abstract OC1.7. European Neuroendocrine Association (ENEA) 14th Congress, 2010:62-63
- U.S. National Library of Medicine: Treatment of pituitary-dependent Cushing’s disease with the multireceptor ligand somatostatin analog pasireotide (SOM230): a multicenter, phase II trial.http://www.ncbi.nlm.nih.gov/pubmed/18957506?dopt=Abstract
- EMEA Approval for Pasireotide
- “FDA Approves Pasireotide for Cushing’s Disease”.
| WO2005117830A1 | 6 Jun 2005 | 15 Dec 2005 | Camurus Ab | Liquid depot formulations |
| WO2006075124A1* | 9 Dec 2005 | 20 Jul 2006 | Camurus Ab | Somatostatin analogue formulations |
| WO2006131730A1 | 6 Jun 2006 | 14 Dec 2006 | Camurus Ab | Glp-1 analogue formulations |
| WO2007096055A1* | 7 Feb 2007 | 30 Aug 2007 | Novartis Ag | Combination of somatostatin-analogs with different selectivity for human somatostatin receptor subtypes |
| WO2010003939A1* | 7 Jul 2009 | 14 Jan 2010 | Novartis Ag | Use of pasireotide for the treatment of endogenous hyperinsulinemic hypoglycemia |
| US20090155193 * | 9 Dec 2005 | 18 Jun 2009 | Fredrik Joabsson | Topical Bioadhesive Formulations |
Read all about Organic Spectroscopy on ORGANIC SPECTROSCOPY INTERNATIONAL 
Join me on Linkedin

Join me on Linkedin



No comments:
Post a Comment