1 AVANAFIL
2 UDENAFIL
3 GISADENAFIL
4 DASANTAFIL
5. Mirodenafil 米罗那非 标准品
6. LODENAFIL CARBONATE
7 VARDENAFIL
8 sildenafil
9 TADALAFIL

watch out...........................

1 AVANAFIL

...............................
4. DASANTAFIL
 Dasantafil
Dasantafil

 DASANTAFIL
DASANTAFIL

 ......................
......................













 DASANTAFIL
DASANTAFIL

 5 Mirodenafil 米罗那非 标准品
5 Mirodenafil 米罗那非 标准品

 MIRODENAFIL米罗那非 标准品
MIRODENAFIL米罗那非 标准品





 methyl salicylate
methyl salicylate
 X=CL, R8=ME
X=CL, R8=ME



 MIRODENAFIL
MIRODENAFIL
.....................................................................................
6 LODENAFIL CARBONATE



 lodenafil
lodenafil
 PIPERAZINE
PIPERAZINE
 ETHYL CHLORO ACETATE
ETHYL CHLORO ACETATE
 Piperazine Ethyl Acetate
Piperazine Ethyl Acetate
![5-(5-Chlorosulfonyl-2-ethoxyphenyl)-1-methyl-3-propyl-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one Structure](https://lh3.googleusercontent.com/blogger_img_proxy/AEn0k_v5QKpf_7zoJe9KKDENz1-AQUhUAu5Rb45qZjXQydH6Dj9vkcEVeczqmWCzIULvUJ1XqoDT-JMrc2s1XzNl8B3jtVYQTsij5ZlyTrhao09_DcuUcGy81w=s0-d)




..............................................................................
7 VARDENAFIL




 LEVITRA
LEVITRA











..........................................................................
8. SILDENAFIL


1-[4-ethoxy-3-(6,7-dihydro-1-methyl-
7-oxo-3-propyl-1H-pyrazolo[4,3-d]pyrimidin-5-yl)
phenylsulfonyl]-4-methylpiperazine
CAS NO 139755-83-2
Sildenafil citrate, sold as Viagra, Revatio and under various other trade names, is a drug used to treat erectile dysfunction and pulmonary arterial hypertension (PAH). It was originally developed by British scientists and then brought to market by the US-based pharmaceutical company Pfizer.[1]It acts by inhibiting cGMP-specific phosphodiesterase type 5 (PDE5), an enzyme that promotes degradation of cGMP, which regulates blood flow in the penis. Since becoming available in 1998, sildenafil has been the prime treatment for erectile dysfunction; its primary competitors on the market are tadalafil (Cialis) and vardenafil (Levitra)


Viagra


SCHEME2

SYNTHESIS

...................................................

...........................................................................
SYNTHESIS

......................................................................................

.................................
PRECURSORS

.........................................
SYNTHESIS

............................................
9 TADALAFIL
























..................................
2 UDENAFIL
3 GISADENAFIL
4 DASANTAFIL
5. Mirodenafil 米罗那非 标准品
6. LODENAFIL CARBONATE
7 VARDENAFIL
8 sildenafil
9 TADALAFIL
watch out...........................
1 AVANAFIL

AVANAFIL, SPEDRA
On 25 April 2013, the Committee for Medicinal Products for Human Use (CHMP) adopted a positive opinion, recommending the granting of a marketing authorisation for the medicinal product Spedra, 50 mg, 100 mg, 200 mg, tablet intended for the treatment of erectile dysfunction in adult men.
The applicant for this medicinal product is VIVUS BV.
They may request a re-examination of any CHMP opinion, provided they notify the European Medicines Agency in writing of their intention within 15 days of receipt of the opinion. The active substance of Spedra is avanafil, a selective phosphodiesterase (PDE) type 5 inhibitor that leads to higher cyclic guanosine monophosphate (cGMP)-specific PDE5 levels. This enhances smooth muscle relaxation, which results in an inflow of blood into the penile tissues, thereby producing an erection. The benefit with Spedra is its effect on the ability of men with erectile dysfunction to achieve and maintain an erection sufficient for satisfactory sexual activity. It was observed in clinical trials that Spedra increased the percentage of sexual attempts resulting in successful intercourse by roughly 20-30% over placebo in the general population of adult men with erectile dysfunction. The most common side effects are headache, flushing, nasal and sinus congestion, dyspepsia and back pain. A pharmacovigilance plan for Spedra will be implemented as part of the marketing authorisation. The approved indication is: "Treatment of erectile dysfunction in adult men. In order for Spedra to be effective, sexual stimulation is required.” Detailed recommendations for the use of this product will be described in the summary of product characteristics (SmPC), which will be published in the European public assessment report (EPAR) and made available in all official European Union languages after the marketing authorisation has been granted by the European Commission. The CHMP, on the basis of quality, safety and efficacy data submitted, considers there to be a favourable benefit-to-risk balance for Spedra and therefore recommends the granting of the marketing authorisation.
Avanafil is a PDE5 inhibitor approved for erectile dysfunction on April 27, 2012.[1] Avanafil is known by the trademark name Stendra and was developed by Vivus Inc. It acts by inhibiting a specific phosphodiesterase type 5 enzyme which is found in various body tissues, but primarily in the corpus cavernosum penis, as well as the retina. Other similar drugs are sildenafil, tadalafil and vardenafil. The advantage of avanafil is that it has very fast onset of action compared with other PDE5 inhibitors.
Avanafil can be synthesized from a benzylamine derivative and a pyrimidine derivative:

"FDA approves Stendra for erectile dysfunction" (Press release). Food and Drug Administration (FDA). April 27, 2012.
Yamada, K.; Matsuki, K.; Omori, K.; Kikkawa, K.; 2004, U.S. Patent 6,797,709
A cutting that phenanthrene by a methylthio urea ( a ) and ethoxy methylene malonate ( 2 ) cyclization of 3 , chloride, phosphorus oxychloride get 4 , 4 with benzyl amine 5 occurred SNAr the reaction product after oxidation with mCPBA 6 . In pyrimidine, if the 2 - and 4 - positions are active simultaneously the same leaving group in the case, SNAr reaction occurs preferentially at 4 - position, but does not guarantee the 2 - side reaction does not occur. Here is an activity of the poor leaving group sulfide spans 2 - bit, and a good leaving group active chlorine occupy four - position, thus ensuring a high regioselectivity of the reaction. 4 - position after completion of the reaction, then the 2 - position of the group activation, where sulfide sulfoxide better than the leaving group. Amino alcohols 7 and 6 recurrence SNAr reaction 8 , 8 after alkaline hydrolysis and acid alpha amidation get that phenanthrene.


 AVANAFIL
AVANAFIL



AVANAFIL
A phosphodiesterase (PDE5) inhibitor, used to treat erectile dysfunction.
Avanafil is a new phosphodiesterase-5 inhibitor that is faster acting and more selective than other drugs belonging to the same class. Chemically, it is a derivative of pyrimidine and is only available as the S-enantiomer. FDA approved on April 27, 2012.
CAS RN: 330784-47-9
4-{[(3-chloro-4-methoxyphenyl)methyl]amino}-2-[(2S)-2-(hydroxymethyl)pyrrolidin-1-yl]-N-(pyrimidin-2-ylmethyl)pyrimidine-5-carboxamide
4-{[(3-chloro-4-methoxyphenyl)methyl]amino}-2-[(2S)-2-(hydroxymethyl)pyrrolidin-1-yl]-N-(pyrimidin-2-ylmethyl)pyrimidine-5-carboxamide
| (S)-2-(2-Hydroxymethyl-1-pyrrolidinyl)-4-(3-chloro-4-methoxybenzylamino)-5-[(2-pyrimidinylmethyl)carbamoyl]pyrimidine |
| 4-[[(3-Chloro-4-methoxyphenyl)methyl]amino]-2-[(2S)-2-(hydroxymethyl)-1-pyrrolidinyl]-N-(2-pyrimidinylmethyl)-5-pyrimidinecarboxamide |
| TA 1790 |
Molecular Formular: C23H26ClN7O3
Molecular Mass: 483.95064
- Stendra
- TA 1790
- TA-1790
- UNII-DR5S136IVO
- NDA 202276
INNOVATOR -- VIVUS
APPROVED FDA 27/4/2-12
| Patent No | Patent Expiry | patent use code |
|---|---|---|
| 6656935 | Sep 13, 2020 | U-155 |
| 7501409 | May 5, 2023 |
U 155... TREATMENT OF ERECTILE DYSFUNCTION
| Exclusivity Code | Exclusivity_Date |
|---|---|
| NCE | Apr 27, 2017 |
Stendra (avanafil) was given the green light by the US Food and Drug Administration 27/4/2012, but there has been no launch yet as Vivus has been seeking a partner. The latest data should be attractive to potential suitors and could help Stendra take on other phosphodiesterase type 5 (PDE5) inhibitors, notably Pfizer’s Viagra (sildenafil) but also Eli Lilly’s Cialis (tadalafil) and Bayer’s Levitra (vardenafil).
read all at
STENDRA (avanafil) is a selective inhibitor of cGMP-specific PDE5.
Avanafil is designated chemically as (S)-4-[(3-Chloro-4-methoxybenzyl)amino]-2-[2-(hydroxymethyl)-1-pyrrolidinyl]-N-(2pyrimidinylmethyl)-5-pyrimidinecarboxamide and has the following structural formula:
 |
Avanafil occurs as white crystalline powder, molecular formula C23H26ClN7O3 and molecular weight of 483.95 and is slightly soluble in ethanol, practically insoluble in water, soluble in 0.1 mol/L hydrochloric acid. STENDRA, for oral administration, is supplied as oval, pale yellow tablets containing 50 mg, 100 mg, or 200 mg avanafil debossed with dosage strengths. In addition to the active ingredient, avanafil, each tablet contains the following inactive ingredients: mannitol, fumaric acid, hydroxypropylcellulose, low substituted hydroxypropylcellulose, calcium carbonate, magnesium stearate, and ferric oxide yellow.
AVANAFIL
Avanafil is a PDE5 inhibitor approved for erectile dysfunction by FDA on April 27, 2012 [1] and by EMA on June 21, 2013.[2] Avanafil is known by the trademark names Stendra and Spedra and was developed by Vivus Inc. In July 2013 Vivus announced partnership with Menarini Group, which will commercialise and promote Spedra in over 40 European countries plus Australia and New Zealand.[3] Avanafil acts by inhibiting a specificphosphodiesterase type 5 enzyme which is found in various body tissues, but primarily in the corpus cavernosum penis, as well as the retina. Other similar drugs are sildenafil, tadalafil and vardenafil. The advantage of avanafil is that it has very fast onset of action compared with other PDE5 inhibitors. It is absorbed quickly, reaching a maximum concentration in about 30–45 minutes.[4] About two-thirds of the participants were able to engage in sexual activity within 15 minutes.[4]
Avanafil is a highly selective PDE5 inhibitor that is a competitive antagonist of cyclic guanosine monophosphate. Specifically, avanafil has a high ratio of inhibiting PDE5 as compared with other PDE subtypes allowing for the drug to be used for ED while minimizing adverse effects. Absorption occurs quickly following oral administration with a median Tmax of 30 to 45 minutes and a terminal elimination half-life of 5 hours. Additionally, it is predominantly metabolized by cytochrome P450 3A4. As such, avanafil should not be co-administered with strong cytochrome P450 3A4 inhibitors. Dosage adjustments are not warranted based on renal function, hepatic function, age or gender. Five clinical trials suggest that avanafil 100 and 200 mg doses are effective in improving the Sexual Encounter Profile and the Erectile Function Domain scores among men as part of the International Index of Erectile Function. A network meta-analysis comparing the PDE5 inhibitors revealed avanafil was less effective on Global Assessment Questionnaire question 1 while safety data indicated no major differences among the different PDE5 inhibitors. The most common adverse effects reported from the clinical trials associated with avanafil were headache, flushing, nasal congestion, nasopharyngitis, sinusitis, and dyspepsia.
A “phosphodiesterase type 5 inhibitor” or “PDE5 inhibitor” refers to an agent that blocks the degradative action of phosphodiesterase type 5 on cyclic GMP in the arterial wall smooth muscle within the lungs and in the smooth muscle cells lining the blood vessels supplying the corpus cavernosum of the penis. PDE5 inhibitors are used for the treatment of pulmonary hypertension and in the treatment of erectile dysfunction. Examples of PDE5 inhibitors include, without limitation, tadalafil, avanafil, lodenafil, mirodenafil, sildenafil citrate, vardenafil and udenafil and pharmaceutically acceptable salts thereof.
“Avanafil” refers to the chemical compound 4-[(3-Chloro-4-methoxybenzyl)amino]-2-[2-(hydroxymethyl)-1-pyrrolidinyl]-N-(2-pyrimidinylmethyl)-5-pyrimidinecarboxamide, and its pharmaceutically acceptable salts. Avanafil is described in Limin M. et al., (2010) Expert Opin Investig Drugs, 19(11):1427-37. Avanafil has the following chemical formula:

Avanafil is being developed for erectile dysfunction. Avanafil currently has no trademarked term associated with it but it is being developed by Vivus Inc.
.......................................
DESCRIPTION IN A PATENT
EXAMPLE 92-145
The corresponding starting compounds are treated in a similar manner to give the compounds as listed in the following Table 7.
| TABLE 7 |
 |
 |  | Amorphous MS(m/z): 484(MH+) |
ENTRY 98 IS AVANAFIL
.............................................................
The invention discloses a preparation method of Avanafil (Avanafil, I), which comprises the following steps: carrying out a substitution reaction on 6-amino-1, 2-dihydro pyrimidine-2-keto-5-carboxylic acid ethyl ester (XII) and 3-chloro-4-methoxy benzyl chloride (XIII) so as to obtain 6-(3-chloro-4-methoxy benzyl amino)-1, 2-dihydro pyrimidine-2-keto-5-carboxylic acid ethyl ester (IXV); carrying out condensation on the compound (IXV) and S-hydroxymethyl pyrrolidine (II) so as to generate 4-[(3-chloro-4-methoxy benzyl) amino]-2-[2-(hydroxymethyl)-1-pyrrole alkyl] pyrimidine-5-carboxylic acid ethyl ester (XI); and carrying out hydrolysis on the compound (XI) and then carrying out an acylation reaction on the compound (XI) and the compound (XI) so as to obtain Avanafil (I). The preparation method is simple in process, economic and environmental-friendly, suitable for the requirements of industrialization amplification.
............................................................
The invention discloses a method for preparing avanafil (Avanafil, I). The method comprises the steps of taking cytosine as an initial material; and orderly carrying out replacement, halogen addition and condensation reaction on a side chain 3-chlorine-4-methoxy benzyl halide (III), N-(2-methylpyrimidine) formamide (IV) and S-hydroxymethyl pyrrolidine (II), so as to obtain a target product avanafil (I). The preparation method is available in material, concise in technology, economic and environment-friendly, and suitable for the demands of industrial amplification.
.............................................................
SYNTHESIS
Avanafil can be synthesized from a benzylamine derivative and a pyrimidine derivative REF 5:Yamada, K.; Matsuki, K.; Omori, K.; Kikkawa, K.; 2004, U.S. Patent 6,797,709
- ...............................................................
- SYNTHESIS
- A cutting that phenanthrene by a methylthio urea ( a ) and ethoxy methylene malonate ( 2 ) cyclization of 3 , chloride, phosphorus oxychloride get 4 , 4 with benzyl amine 5 occurred SNAr the reaction product after oxidation with mCPBA 6 . In pyrimidine, if the 2 – and 4 – positions are active simultaneously the same leaving group in the case, SNAr reaction occurs preferentially at 4 – position, but does not guarantee the 2 – side reaction does not occur. Here is an activity of the poor leaving group sulfide spans 2 – bit, and a good leaving group active chlorine occupy four – position, thus ensuring a high regioselectivity of the reaction. 4 – position after completion of the reaction, then the 2 – position of the group activation, where sulfide sulfoxide better than the leaving group. Amino alcohols 7 and 6 recurrence SNAr reaction 8 , 8 after alkaline hydrolysis and acid alpha amidation get that phenanthrene.
AVANAFIL
- ..................................

- FDA approves Stendra for erectile dysfunction" (Press release). Food and Drug Administration (FDA). April 27, 2012.
- http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/002581/human_med_001661.jsp&mid=WC0b01ac058001d124
- http://ir.vivus.com/releasedetail.cfm?releaseid=775706
- Kyle, Jeffery; Brown, Dana (2013). "Avanafil for Erectile Dysfunction". Annals of Pharmacotherapy (Sage Publishing). doi:10.1177/1060028013501989. Retrieved 28 September 2013.
- Yamada, K.; Matsuki, K.; Omori, K.; Kikkawa, K.; 2004, U.S. Patent 6,797,709
- Peterson CA. Hemodynamic effect of avanafil and glyceryl trinitrate coadministration. , Drugs Context , Volume 2013 , 2013 Feb 26
- Gur S. The Effect of Intracavernosal Avanafil, a Newer Phosphodiesterase-5 Inhibitor, on Neonatal Type 2 Diabetic Rats With Erectile Dysfunction. , Urology , 2013 Dec 9
- Hill JK. Avanafil for erectile dysfunction. , Ann Pharmacother , Volume 47 , Issue 10 , 2013 Oct
- Sanford M. Avanafil: a review of its use in patients with erectile dysfunction. , Drugs Aging , Volume 30 , Issue 10 , 2013 Oct
- Hellstrom WJ. PDE5 inhibitors: considerations for preference and long-term adherence. , Int J Clin Pract , Volume 67 , Issue 8 , 2013 Aug
- Aversa A. An update on pharmacological treatment of erectile dysfunction with phosphodiesterase type 5 inhibitors. , Expert Opin Pharmacother , Volume 14 , Issue 10 , 2013 Jul
- Oelke M. Phosphodiesterase inhibitors in clinical urology. , Expert Rev Clin Pharmacol , Volume 6 , Issue 3 , 2013 May
- Kukreja RC. Sildenafil and cardioprotection. , Curr Pharm Des , Volume 19 , Issue 39 , 2013
- Day WW. An open-label, long-term evaluation of the safety, efficacy and tolerability of avanafil in male patients with mild to severe erectile dysfunction. , Int J Clin Pract, Volume 67 , Issue 4 , 2013 Apr
- Tang J. Comparative effectiveness and safety of oral phosphodiesterase type 5 inhibitors for erectile dysfunction: a systematic review and network meta-analysis. , Eur Urol , Volume 63 , Issue 5 , 2013 May
United States APPROVED 6656935 2012-04-27 EXPIRY 2020-09-13 United States 7501409 2012-04-27 2023-05-05 - Faster-Working Erectile Dysfunction Drug?. CBS News. November 24, 2009.
- Vivus says men taking avanafil were more likely to be ready for sex within 15 minutes. The Gaea Times. January 11, 2010.
- "Avanafil is the New Player in The Erectile Dysfunction Field". June 28, 2011.
- • Hatzimouratidis, K., et al.: Drugs, 68, 231 (2008)
US7927623 4-20-2011 Tablets quickly disintegrated in oral cavity US2010179131 7-16-2010 Combination treatment for diabetes mellitus US2009215836 8-28-2009 Roflumilast for the Treatment of Pulmonary Hypertension US2008027037 1-32-2008 Cyclic compounds
US5242391 Oct 30, 1991 Sep 7, 1993 ALZA Corporation Urethral insert for treatment of erectile dysfunction US5474535 Jul 19, 1993 Dec 12, 1995 Vivus, Inc. Dosage and inserter for treatment of erectile dysfunction US5773020 Oct 28, 1997 Jun 30, 1998 Vivus, Inc. Treatment of erectile dysfunction US6656935 Aug 10, 2001 Dec 2, 2003 Tanabe Seiyaku Co., Ltd. Aromatic nitrogen-containing 6-membered cyclic compounds
......................................
2 UDENAFIL
UDENAFILAn oral phosphodiesterase 5 inhibitor used for the treatment of erectile dysfunction.268203-93-6 CAS NOLAUNCHED 2005 MEZZION DA-8159 ME-3113 Udzire Zydena MEZZION ...INNOVATORSynonyms: Zydena;Udenafi;Da-8159;Da 8159;Udenafil;Udenafil(DA 8159,Zydena);5-(2-Propyloxy-5-(1-methyl-2-pyrollidinylethylamidosulfonyl)phenyl)-1-methyl-3-propyl-1,6-dihydro-7H-pyrazolo(4,3-D)pyrimidine-7-one;5-[2-propyloxy-5-[2-(1-Methyl-2-pyrrolidinyl)ethylaMinosulfonyl]phenyl]-1-Methyl-3-propyl-1,6-dihydro-7H-pyrazolo[4,3-d]pyriMidine-7-one;5-[2-propyloxy-5-(2-(1-Methylpyrrolidin-2-yl)ethylaMinosulphonyl)phenyl]-1-Methyl-3-propyl-6,7-dihydro-1H-pyrazolo(4,3-d)pyriMidin-7-one;3-(6,7-Dihydro-1-methyl-7-oxo-3-propyl-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-N-[2-(1-methyl-2-pyrrolidinyl)ethyl]-4-propoxybenzenesulfonamide Molecular Formula: C25H36N6O3S2 Formula Weight: 516.66 3-(1-methyl-7-oxo-3-propyl-4,7-dihydro-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-N-[2-(1-methylpyrrolidin-2-yl)ethyl]-4-propoxybenzenesulfonamide(5- [2-propyloxy-5- (1- methyl-2-pyrolidinylethylamidosulfonyl) phenyl] -1-methyl- propyl-1, β-dihydro-7H-pyrazolo (4 , 3-d) pyrimidin-7-one)A pyrazolo-pyrimidinone similar to sildenafil; phosphodiesterase type 5 inhibitor. Udenafil is a new phosphodiesterase type 5 (PDE5) inhibitor used to treat erectile dysfunction (ED). It has been approved in South Korea and will be marketed under the brand name Zydena.It is not yet approved for use in the U.S., E.U., or Canada. Udenafil (Zydena®) is also a potent and selective PDE5i developed by Dong-A Pharmaceutical Company in Korea (Kim et al., 2008; Han et al., 2010).It has not yet been approved by FDA or the European Medicines Agency (EMEA) and was only approved by the Korean Food and Drug Administration (KFDA), being currently used in Korea and Russia (Alwaal et al., 2011; Cho et al., 2012).- DA 8159
- DA-8159
- Udenafil
- UNII-L5IB4XLY36
- Zydena
Udenafil is a drug used in urology to treat erectile dysfunction. It belongs to a class of drugs called PDE5 inhibitor, which many other erectile dysfunction drugs such as sildenafil, tadalafil, and vardenafil also belong to. It was developed by Dong-A Pharmaceutical Co., Ltd. and is marketed under the trade name Zydena™.[2] With a T max of 1.0-1.5 h and a T 1/2 of 11-13 h (a relatively rapid onset and a long duration of action), both on-demand and once-daily use of udenafil have been reported.[3] Typical doses are 100 and 200 mg. It is not approved for use in the United States by theU.S. Food and Drug Administration. Udenafil (DA-8159), a pyrazolopyramidinone derivative that acts as a phosphodiesterase 5 (PDE5) inhibitor, was launched by Dong-A Pharmtech (currently Mezzion Pharma) in late 2005 in Korea for the oral treatment of erectile dysfunction (ED). The company is currently conducting phase III clinical trials in the U.S. for this indication.Dong-A Pharmatech is conducting phase III clinical trials for the treatment of patients with portal hypertension resulting from liver disease and for the treatment of benign prostatic hyperplasia (BPH). Phase II/III clinical studies at Dong-A Pharmatech for the treatment of secondary Raynaud phenomenon have been completed. Meiji Seika Pharma is developing the compound in phase I clinical trials for the treatment of BPH in Japan.Phosphodiesterases regulate the tissue concentration of cyclic guanosine monophosphate (cGMP), which in turn triggers smooth muscle relaxation, allowing blood to flow into the penis and resulting in erection. PDE5 is the most abundant phosphodiesterase in the human corpus cavernosum, and as such its inhibition by DA-8159 enhances erectile function by increasing the concentration of cGMP. Results from phase I studies indicate that udenafil has a unique pharmacokinetic profile with a relatively rapid onset and sufficiently long duration to make it effective for up to 24 hours. In 2009, the compound was licensed to Warner Chilcott (acquired by Actavis in 2013) by Dong-A Pharmatech for development and marketing in the U.S. for the oral treatment of erectile dysfunction.In 2011, udenafil was licensed to Meiji Seika Pharma by Dong-A ST in Japan for the treatment of benign prostatic hyperplasia. Udenafil is a potent novel phosphodiesterase-5 inhibitor approved for use in Korea. Udenafil has unique properties, with a T max of 1.0–1.5 h and a T 1/2 of 11–13 h (a relatively rapid onset and a long duration of action). Therefore, both on-demand and once-daily use of udenafil have been reported. Udenafil’s efficacy and tolerability have been evaluated in several studies, and recent and continuing studies have demonstrated udenafil’s promise in both dosing regimens. Presently, tadalafil is the only FDA-approved drug for daily dosing, but udenafil can be used as a once-daily dose for erectile dysfunction patients who cannot tolerate tadalafil due to phosphodiesterase subtype selectivity. Udenafil as an on-demand or once-daily dose is effective and tolerable, but more studies are needed in patients of other ethnicities and with comorbid conditions such as diabetes mellitus, hypertension, and benign prostate hyperplasia.Erectile dysfunction (ED) is defined as the inability to achieve and maintain a sufficient erection to permit satisfactory intercourse [Montorsi et al. 2010]. Numerous strategies have been used to overcome ED. Therapies for ED include intracavernosal injection, vacuum erection devices, intraurethral suppositories, penile prosthesis surgery and oral phosphodiesterase-5 (PDE5) inhibitors [Dinsmore and Evans, 1999]. Oral PDE5-inhibitor medications have revolutionized the treatment of ED. Men prefer oral medications as the first-line therapeutic option in the absence of a specific contraindication to their use [Ding et al. 2012].There are currently four PDE5 inhibitors (sildenafil, vardenafil, tadalafil, and avanafil) approved worldwide for the treatment of male erectile dysfunction, with two other agents (udenafil and mirodenafil) currently approved only in Korea [Bell and Palmer, 2011]. The choice of PDE5 inhibitor for each patient should be determined after physician and patient discuss the characteristics of different drugs and the individual patient’s sexual habits, preferences, and expectations [Hatzimouratidis et al. 2010]. There are two types of treatment usage of PDE5 inhibitors according to their pharmacological characteristics. On-demand treatment of ED with PDE5 inhibitors allows the patient to have intercourse within 1 hour, but can remove spontaneity from sexual activity and be burdensome to patients and their partners [Hanson-Divers et al. 1998]. Once-daily dosing of a PDE5 inhibitor is an alternative for couples that prefer spontaneous sexual activities.A new oral selective PDE5 inhibitor, udenafil (Zydena, Dong-A, Seoul, Korea), has recently been developed for the treatment of ED. Udenafil is a novel pyrazolopyrimidinone compound developed by Dong-A Pharmaceutical Co., Ltd (Seoul, Korea) for the treatment of ED which has the same mechanism of action as sildenafil [Kim et al. 2008]. Udenafil is rapidly absorbed, reaching peak plasma concentrations at 0.8–1.3 h, then declining monoexponentially with a terminal half-life (T 1/2) between 7.3 and 12.1 hours, giving it the unique pharmacokinetics of both relatively rapid onset and long duration [Salem et al. 2006]. Thus, both on-demand treatment and once-daily dosing have been reported in the literature. The purpose of this review is to evaluate the efficacy and tolerability of udenafil for patients with ED according to the currently available literature.Udenafil” refers to the chemical compound, 3-(1-methyl-7-oxo-3-propyl-4,7-dihydro-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-N-[2-(1-methylpyrrolidin-2-yl)ethyl]-4-propoxybenzenesulfonamide and has the following chemical formula:More information about udenafil can be found at Kouvelas D. et al., (2009) Curr Pharm Des, 15(30):3464-75. Udenafil is marketed under the trade name Zydena® but not approved for use in the United States. TRADE NAME IN INDIA – UDEZIRE Erectile dysfunction (ED) is an inability to achieve or sustain an erection suitablefor sexual intercourse.Sexologists say at least 10% men in India may have to use these drugs at some point. Studies have shown that 40% of men up to the age of 40 years have ED andit goesup 70% by 60 years. The commonly prescribed drugs for the disorder in India are sildenafil(Viagra) and tadalafil,which belong to a category called phosphodiesterasetype5 drugs.Now, Zydus, a pharmaceutical company, has got exclusive permission to sell udenafil. It’s not always that the release of a drug is celebrated by many, particularly men. A drug that was released in India last week is the recent in the list of drugs that has a cure for erectile dysfunction. The manufacturers say udenafil, which will be marketed under the brand name Udezire, will be long-acting, but with minimal side effects. Erectile dysfunction (ED) is an inability to achieve or sustain an erection suitable for sexual intercourse. Sexologists say at least 10% men in India may have to use these drugs at some point. Studies have shown that 40% of men up to the age of 40 years have ED and it goes up 70% by 60 years
Udenafil like Sildenafil, Tadalafil, Avanafil, and Vardenafil (Viagra®, Cialis®, Stendra ® and Levitra® respectively) is an orally taken PDE-5 inhibitor. Its function is very similar in that it blocks the action of phosphodiesterase type 5 and relieves erectile dysfunction in men.Udenafil is produced by Dong-A PharmTech Co Ltd. from Korea and has actually been used there since Nov 2005 and marketed as Zydena® and has since been approved for use in Russia in 2008. An indication that it may indeed prove to be a factor in the ED medication mix in the US one day. In 2009 Dong-A Pharmaceutical Co., Ltd. and Dong-A PharmTech, Co. Ltd. announced that it had completed a 240 patient once-a-day dosing clinical study of udenafil, its new long acting phosphodiesterase type 5 (PDE-5) inhibitor for erectile dysfunction (ED).The multi-center study conducted in Korea was a randomized, double-blind, placebo-controlled study, designed to investigate the efficacy and safety of udenafil in patients with ED. Following a 4-week non-drug baseline period, 240 men with ED of broad etiology and severity were randomized to one of four treatment groups: Placebo, udenafil 25 mg, udenafil 50 mg or udenafil 75 mg. Patients took one tablet a day for 12 weeks with evaluations every 4 weeks.The primary efficacy endpoint was the change in the standard International Index of Erectile Function (IIEF) Erectile Function Domain (EF) score from baseline to final visit. The secondary efficacy endpoints were the change from the baseline in the mean vaginal penetration success rates and mean intercourse completion rates calculated from the Sexual Encounter Profile (SEP) questions 2 and 3. In addition a sub-group analysis was conducted to determine efficacy in the patients that had lower urinary tract symptoms associated with benign prostatic hyperplasia in addition to erectile dysfunction.UDENAFIL
........................INTRODUCTIONUdenafil (Zydena®) is a therapeutic agent hypothesized to improve erectile function endpoints through interaction with the phosphodiesterase type 5 (PDE5) enzyme. As such, udenafil belongs to the class of such agents that includes tadalafil (Clalis®), sildenafil (Viagra®), and vardenafil (Levitra®). These agents are purported to promote erectile response through inhibition of PDE5, the predominant PDE within the penis, which leads to higher intracellular levels of cyclic guanylate cyclase (cGMP). cGMP is a second messenger for the smooth-muscle relaxing effects of nitric oxide within the penis. The various agents differ in pharmacology primarily based on 1) onset and duration of action and 2) selectivity profiles vs. other PDEs. All three marketed agents have proven remarkably safe. These agents should not be taken by patients with unstable cardiovascular disease. Udenafil has been shown to exhibit greater selectivity against the known PDE homologues, than other PDE5 inhibitors. Udenafil is comparable to tadalafil in many respects, such as duration of action and high selectivity for PDE6, but udenafil has greater selectivity for PDE11 than tadalafil.Tadalafil, with a half life of 17.5 hours, has a much longer duration of action and improved exercise tolerance than either sildenfail or vardenafil, which have half lives of 4-5 hours. Consequently, tadalafil is associated with less planning or pressure to have sexual intercourse after dosing. Dissociation of the sexual activity from the time of dosing is associated with higher rates of patient and partner satisfaction. In prospective, randomized crossover clinical studies, patients preferred tadalafil over sildenafil by margins ranging from 7:3 to 9:1. Sildenafil and vardenafil both modulate PDE6 at higher rate than tadalafil. PDE6 modulation has been associated with chromatopsia. The side effects of chromatopsia, such as sensitivity to light and blurred vision, are therefore higher in patients taking sildenafil or vardenafil, about 2-3%, than patients taking tadalafil, about <0.1%. Tadalafil is less selective than sildenafil and vardenafil for PDE5 and for PDE11a. Activity at PDE11a is suspected to have a causal relationship with myalgia and testicular toxicity. The selectivity profile for udenafil is similar to sildenafil, which should impart greater safety for this agent.The benefits and shortcomings of these drugs have been reviewed. Some of these shortcomings can be traced to metabolism-related phenomena. Udenafil is converted in vivo by oxidative and conjugative degradation to multiple metabolites. Phase I metabolism leads to demethylation of the pyrazole, hydroxylation of the pyrazole propyl group, and dealkylation alpha to the sulfonamide nitrogen to afford an active metabolite. Because udenafil is metabolized primarily by cytochrome P450 subtype 3A4 (CYP3A4), exposure to udenafil can influence polypharmacy. For example, CYP3A4 inhibitors such as HIV protease inhibitors, azole antifungals, and erythromycin can lead to higher than otherwise expected blood levels of udenafil. Conversely, co-administration of CYP3A4 inducers such as rifampin can decrease the otherwise expected blood levels of udenafil. Thus, the polypharmacy of udenafil is necessarily complex and has potential for adverse events. In addition, there may be increased inter-patient variability in response to polypharmacy.Analogs of udenafil as described herein have the potential to alleviate the problems associated with the commercially available PDE5 inhibitors while maintaining or improving efficacy. It is believed that the reduction in CYP3A4 clearance of udenafil analogs will be expected to increase the proportion of clearance via mechanisms less susceptible to polypharmaceutical complications. In addition, analogs of udenafil having an attenuated rate of oxidative metabolism will have an increased half-life, further augmenting their advantages vs. tadalafil, sildenafil and vardenafil. Potentially, a single dose of an udalafil analog, described herein, having an increased half-life may provide therapeutic coverage for an entire weekend or beyond while increasing safety parameters by reducing the likelihood of drug-drug interactions and by increasing safety as a result of the increased selectivity.The compounds of formula 1 may contain asymmetric centers and thus they can exist as enantiomers. The present invention includes both mixtures and separate individual isomers . Male erectile dysfunction is one of the most common sexual dysfunctions in men. Although erectile dysfunction can be primarily psychogenic in origin, it often accompanies chronic illnesses, such as diabetes mellitus, heart disease, hypertension, and a variety of neurological diseases. Its prevalence is strongly related to age, with a estimated prevalence of 2% at age 40 years rising to 25-30% by age of 65. Although no data are available on the prevalence of erectile dysfunction in men aged over 75, it is probably over 50%. Various treatment options for erectile dysfunction are available, such as counseling, hormonal therapy, self-injection or transurethral application of vasodilator agents, vacuum devices, prosthesis implantation, and venous/arterial surgery. However, these therapeutic options have several limitations such as side effects, high-cost and low efficacy.Therefore it has called for research efforts to develop new, high effective and simple to use treatment methods, potentially oral medication. Recently, sildenafil has been developed as a therapeutic agent for male erectile dysfunction by oral administration. Sildenafil is the first in a new class of drugs known as inhibiting phosphodiesterase-5 enzyme distributed specifically in corpus cavernosal tissues and induces relaxation of the corpus cavernosal smooth muscle cells, so that blood flow to the penis is enhanced, leading to an erection.Sildenafil has shown a response rate of around 80% in men with erectile dysfunction of organic cause. On the other hand, USP 3,939,161 discloses that 1 , 3 -dimethyl -lH-pyrazolopyrimidinone derivatives exhibit anticonvulsant and sedative activiity, and also exhibit anti-inflammatory activity and gastric antisecretory activity; EP 201,188 discloses that 5-substituted pyrazolopyrimidinone derivatives have effects of antagonizing adenosine receptor and of inhibiting phosphodiesterase enzymes and can be used for the treatment of cardiovascular disorders such as heart failure or cardiac insufficiency; EP 463,756, EP 526,004, WO 93/6,104 and WO 93/7,149 disclose that pyrazolopyrimidinone derivatives which inhibit c-GMP phosphodiesterase more selectively than c-AMP phosphodiesterase have efficacy on cardiovascular disorders such as angina pectoris, hypertension, heart failure, atherosclerosis, chronic asthma, etc.; and WO 94/28,902, WO 96/16,644, WO 94/16,657 and WO 98/49,166 disclose that the known inhibitors of c-GMP phosphodiesterase including the pyrazolopyrimidinone derivatives of the above mentioned patents can be used for the treatment of male erectile dysfunction Since sildenafil has been developed, various compounds for inhibiting phosphodiesterase-5 have been reported.Among them, pyrazolopyrimidinone compounds of formula 1 (KR Pat. No. 99-49384) were reported having better potency than that of sildenafil, based on the mechanism of inhibiting phosphodiesterase-5 and having better selectivity over phosphodiesterase-6 distributed in retina and phosphodiesterase-3 distributed in heart to reduce the side effects. Further, the pyrazolopyrimidinone compounds of formula 1 were said to be improved the solubility and the metabolism in the liver, which are very important factor affecting the rate of the absorption when administered orally.The KR patent No. 99-49384 also disclosed a process for preparing the pyrazolopyrimidinone compounds of formula , comprising the steps of: a) reacting chlorosulfonated alkoxy bonzoic acid with a primary amine to obtain sulfonamide-substituted benzoic acid; b) reacting the obtained sulfonamide-substituted benzoic acid with pyrazolamine in the presence of activating reagent of carboxylic group or coupling agent of carboxylic group with amine group to obtain corresponding amide compound; and, c) performing an intramolecular cyclization of the obtained amide compound to obtain the pyrazolopyrimidinone compound of formula 1. This reaction is represented in scheme 1 Scheme 1.......................SYNTHESISThe present invention provides an agent comprising a pyrazolopyrimidinone compound (5- [2-propyloxy-5- (1- methyl-2-pyrolidinylethylamidosulfonyl) phenyl] -1-methyl- propyl-1, 6-dihydro-7H-pyrazolo (4, 3-d) pyrimidin-7-one) expressed as formula 1 as an effective ingredient for preventing and treating benign prostatic hyperplasia (BPH) . Formula 1The pyrazolopyrimidinone compound represented as formula 1 is one of the PDE-5 inhibitors and has characteristics in that it has a strong inhibitive activity and an excellent selectivity for PDE-5; it is readily absorbed as its solubility is improved; it has a good bioavailability and a large volume of distribution; and it has an in vivo half-life longer three times than sildenafil or vardenafil, a drug of the same mechanism. Physicochemical properties of the pyrazolopyrimidinone compound of formula 1 are as follows: it is hardly dissolved in water; however, it is readily dissolved in acetic acid, methanol, chloroform and the like; and it is a white or pale yellow powder, not a hydrate or a solvate, having a melting point of 158 to 161 "Q and having pKal and pKa2 of about 6.5 and 12.5, respectively. The pyrazolopyrimidinone compound represented as formula 1 is prepared via a synthetic process consisting of roughly three steps. The inventors of the present invention have disclosed a method for preparing the same in WO2000/027847 (Corresponding Korean Patent No.0353014), which will now be described roughly as follows. First, in the first step, 4- [2-propyloxy-5- (chlorosulfonyl) benzamido] -l-methyl-3-propyl-5-carbamoyl pyrazole is prepared.For such preparation, a specified amount of 4- [2-propyloxybenzamido] -l-methyl-3-propyl-5- carbamoyl pyrazole is added to a specified amount of chlorosulfonic acid cooled to 0 °Q then, the resultant mixture is stirred, filtered, washed and dried to obtain 4- [2-propyloxy-5- (chlorosulfonyl) benzamido] -l-methyl-3- propyl-5-carbomoyl pyrazole. In the second step, from the pyrazole compound prepared in the first step, 4- [2-propyloxy-5- ( l-methyl-2- pyrolidinylethylamidosulfonyl) benzamido] -l-methyl-3- propyl-5-carbomoyl pyrazole is prepared. For such preparation, a specified amount of 2- (2-aminoethyl) -1- methyl pyrolidine is added in dichloromethane solution of the specified amount of 4- [2-propyloxy-5- (chlorosulfonyl) benzamido] -l-methyl-3-propyl-5-carbamoyl pyrazole prepared in the first step to be stirred. Then, the reactant solution is diluted with dichloromethane. The organic layer is washed, dried, concentrated and filtered to obtain 4- [2-propyloxy-5- (l-methyl-2- pyrolidinylethylamidosulfonyl) benzamido] -l-methyl-3- propyl-5-carbomoyl pyrazole is obtained.Last, in the third step, the pyrazolopyrimidinone compound of the present invention (5- [2-propyloxy-5- (1- methyl-2-pyrolidinylethylamidosulfonyl) phenyl] -1-methyl- propyl-1, β-dihydro-7H-pyrazolo (4 , 3-d) pyrimidin-7-one) is prepared from the compound obtained in the second step. For such preparation, the specified amount of pyrazole compound prepared in the second step is dissolved in t- butanol . A specified amount of potassium t-butoxide is added in the resultant solution and, then, reflux-stirred for a predetermined time. After the resultant solution is cooled, diluted, washed and dried, distillation under reduced pressure, solvolysis and silica gel column chromatography are carried out, thus obtaining a specified amount of pure pyrazolopyrimidinone compound of the present invention. ................................SYNTHESIS WO2000027848A1REACTION SCHEME 2The process for preparation according to the present invention comprises the steps of : 1) reacting the chlorosulfonated compound of formula ( 2 ) and primary amine (3_) under the condition of suitable temperature and suitable solvent to give sulfonamide (4.) (step 1) ; 2) reacting the carboxylic acid (4.) prepared in step 1 and pyrazoleamine (5) to give an amide (6.) by the known method preparing amide from carboxylic acid and amine (step 2) ; and 3) cyclizing the amide (6.) prepared in step 2 to give the desired compound of formula 1 by the known cyclization method used for preparation of pyrimidinone (step 3) .In step 1, a little excess of 2 equivalents of amine may be used, or a little excess of 1 equivalent of amine and 1 equivalent of acid scavenger such as tertiary amine are may be used together. The reaction temperature is preferred below 20 °C. The known method preparing amide from carboxylic acid and amine in step 2 is the process, for example, in which carboxyl group is transformed into activated acid chloride or acid anhydride by using thionyl chloride, pivaloyl chloride, trichlorobenzoyl chloride, carbonyldiimidazole, diphenylphosphinic chloride, etc. and followed by reacting with amine group, or the process using coupling agents such as DCC (1,3-dicyclo hexylcarbodiimide) or EEDQ (N-ethoxycarbonyl -2 -ethoxy- 1, 3-dihydroquinoline) .The cyclization process in step 3 may be carried out in the presence of a suitable base and a suitable solvent. Preferred bases which are employed in step 3 are metal alkoxides; metal salts of ammonia; amine; hydrides of alkali metal or alkaline earth metal; hydroxides; carbonates; bicarbonates ; and bicyclic amidines such as DBU (1 , 8-diazabicyclo [5.4.0] undec -7-ene) and DBΝ (1 , 5-diazabicyclo [4.3.0] non-5-ene) . Preferred solvents which are employed in step 3 are alcohols such as methanol, ethanol, isopropanol, t-butanol, etc.; ethers such as tetrahydrofuran, dimethoxyethane, dioxane, etc.; aromatic - hydrocarbons such as benzene, toluene, xylene, chlorobenzene, etc.; acetonitrile; dimethylsulfoxide; dimethylformamide; N-methylpyrrolidin-2 -one ; and pyridine.SEE ENTRY no 685- [2-propyloxy-5- ( 1-methyl-2-pyrrolidinylethyl amidosulfonyl) phenyl] -l-methyl-3 -propyl-1 , 6-dihydro-7 H-pyrazolo (4 , 3-d) yrimidin-7-one (compound of example68)ACCORDING TO ME ENTRY IS 68 ANY ERROR, amcrasto@gmail.comSynthesis WO2001098304A1The present invention relates to a process for preparing pyrazolopyrimidinone derivatives of formula 1 and pharmaceutically acceptable salts thereof which have an efficacy on impotence, comprising the steps of chlorosulfonation of pyrazolamide compounds of formula 2, followed by amination with a primary amine and intramolecular cyclization. Formula 1Formula 2The compounds of formula 1 may exist in tautomeric equilibrium as shown below.The compounds of formula 1 may also contain asymmetric centers and thus they can exist as enantiomers. The present invention includes both racemic mixture and separate individual enantiomers. Scheme 2.....................................SYNTHESIS WO2010013925A2INTERMEDIATES4-[2-propyloxy benzamido]-l-methyl-3-propyl-5-carbamoyl pyrazoleCHLOROSULPHONIC ACID4-[2-propyloxy-5-(chlorosulfonyl)benzamido]-l-methyl-3-propyl-5-carbamoyl pyrazole2-(2-aminoethyl)-l-methylpyrrolidine 4-[2-propyloxy-5-(l-methyl-2-pyrrolidinylethyl amido- sulfonyl)benzamido]-l-methyl-3-propyl-5-carbamoyl pyrazolepotassium t-butoxide3, 5-[2-propyloxy-5-(l-methyl-2-pyrrolidinylethyl amido- sulfonyl)phenyl]-l-methyl-3-propyl-l,6-dihydro-7H-pyrazolo(4,3-d)pyrimidin-7-one UDENAFILThe present invention provides a pharmacological compound containing 5- [2-propyloxy-5-( 1 -methyl-2-pyrolidinylethylamidosulphonyl)phenyl] - 1 -methyl-prop yl- 1 ,6-dihydro-7H-pyrazolo(4,3-d)pyrimidin-7-one, a pyrazolopyrimidinone compound, represented by the following Chemical Formula 1 or pharmaceutically acceptable salts thereof, as an active ingredient for prevention and treatment of respiratory diseases. [14] [Chemical Formula 1]Best Mode for Carrying out the Invention [26] The pyrazolopyrimidinone compound of Chemical Formula 1 is a kind of phosphodiesterase type 5 inhibitor. The compound has excellent PDE 5 inhibitory activity and selectivity. It is absorbed fast due to its improved solubility, and has high bioavailability and huge volume of distribution. It is characterized by about a 3-fold longer elimination half- life than those of sildenafil or vardenafil, drugs with the same mechanism.[27] The pyrazolopyrimidinone compound of Chemical Formula 1 is not a hydrate or solvate, but a white or light-white powder with the melting point of 158-1610C and the pKal and pKa2 values of about 6.5 and 12.5, respectively. The compound is insoluble in water, but soluble in acetic acid, methanol, and chloroform.[28] The pyrazolopyrimidinone compound of Chemical Formula 1 is prepared through a three-step synthetic process and a preparation method of the compound is disclosed in WO 00/027848 and KR Patent No. 0353014. The method is briefly described as follows.[29] In Step 1, 4-[2-propyloxy-5-(chlorosulfonyl)benzamido]-l-methyl-3-propyl-5-carbamoyl pyrazole is prepared. For preparation, a predetermined amount of 4-[2-propyloxy benzamido]-l-methyl-3-propyl-5-carbamoyl pyrazole is added to a predetermined amount of chlorosulfonic acid cooled at O0C. The reaction mixture is stirred, filtered, washed and dried to obtain 4-[2-propyloxy-5-(chlorosulfonyl)benzamido]-l-methyl-3-propyl-5-carbamoyl pyrazole.[30] In Step 2, 4-[2-propyloxy-5-(l-methyl-2-pyrrolidinylethyl amido- sulfonyl)benzamido]-l-methyl-3-propyl-5-carbamoyl pyrazole is prepared from the pyrazole compound prepared in the above step 1. For preparation, a predetermined amount of 2-(2-aminoethyl)-l-methylpyrrolidine is added at O0C to a dichloromethane solution containing a predetermined amount of 4-[2-propyloxy-5-(chlorosulfonyl)benzamido]-l-methyl-3-propyl-5-carbamoyl pyrazole of step 1, followed by stirring. Upon completion of the reaction, the reaction solution is diluted with dichloromethane. The organic layer is washed, dried, concentrated and filtered to obtain 4-[2-propyloxy-5-(l-methyl-2-pyrrolidinylethyl amido- sulfonyl)benzamido]- l-methyl-3-propyl-5-carbamoyl pyrazole.[31] In step 3, 5-[2-propyloxy-5-(l-methyl-2-pyrrolidinylethyl amido- sulfonyl)phenyl]-l-methyl-3-propyl-l,6-dihydro-7H-pyrazolo(4,3-d)pyrimidin-7-one, UDENAFIL a pyrazolopyrimidinone compound of the present invention, is prepared from the compound obtained in step 2. For preparation, a predetermined amount of the pyrazole compound synthesized in step 2 is dissolved in t-butanol, to which a predetermined amount of potassium t-butoxide is added, followed by stirring under reflux for a predetermined time. Upon completion of the reaction, the reaction solution is cooled down, diluted, washed and dried. Then, reduced pressure distillation, elimination of a solvent and silica gel column chromatography are performed to obtain a predetermined amount of a novel pyrazolopyrimidinone compound of the invention, represented by Chemical Formula 1...................SYNTHESISEXAMPLE 2 3-(1-Methyl-7-oxo-3-propyl-6,7-dihydro-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-N-[2-(1-methyl-pyrrolidin-2-yl)-ethyl]-4-propoxy-benzenesulfonamideStep 12,4-Dioxo-heptanoic acid methyl ester: Sodium (25.3 g, 1.1 mol) was proportionally added to ethanol (350 mL) at ambient temperature with vigorous stirring, and the solution was cooled to 0° C. Pentan-2-one (86 g, 1.0 mol) and diethyl oxalate (146 g, 1.0 mol) were added sequentially at 0° C., and stirring was continued for 1 hour at 0° C., and overnight at ambient temperature. The solvent was removed under reduced pressure, diethyl ether (200 mL) and cold dilute hydrochloric acid (500 mL) were added. Following standard extractive work up, the solvent was evaporated under reduced pressure to yield the title compound (141 g, 76%). 1H-NMR (300 MHz, CDCl3) δ 14.51 (broad s, 1H), 6.37 (s, 1H), 4.35 (q, 2H, J=6.6 Hz), 2.47 (t, 2H, J=7.2 Hz), 1.76-1.66 (m, 2H), 1.38 (t, 3H, J=7.2 Hz), 0.97 (t, 3H, J=7.5 Hz); GC-MS: 186 (M)+, 113 (M-73)+Step 25-Propyl-2H-pyrazole-3-carboxylic acid ethyl ester: Hydrazine hydrate (41.4 g, 827 mmol) was slowly added to a solution of 2,4-dioxo-heptanoic acid methyl ester (140 g, 753 mmol) in 280 mL of acetic acid at 0° C. The mixture was heated to reflux for 8 hours and cooled. The solvent was removed under reduced pressure; the residue was diluted with diethyl ether (300 mL). Following standard extractive work up, the solvent was evaporated under reduced pressure to yield the title compound as a white solid (131 g, 96%). 1H NMR (300 MHz, CDCl3) δ 9.27 (broad s, 1H), 6.61 (s, 1H), 4.37 (q, 2H, J=7.2 Hz), 2.68 (t, 2H, J=7.5 Hz), 1.75-1.62 (m, 2H), 1.37 (t, 3H, J=6.6 Hz), 0.96 (t, 3H, J=7.2 Hz); LC-MS: m/z=183 (MH)+;Step 32-Methyl-5-propyl-2H-pyrazole-3-carboxylic acid ethyl ester: A mixture of 5-propyl-2H-pyrazole-3-carboxylic acid ethyl ester (32.8 g, 180 mmol) and dimethyl sulfate (24.9 g, 198 mmol) was heated at 90° C. for 3 hours. The reaction was cooled and diluted with dichloromethane (200 mL). Following standard extractive work up, the solvent was evaporated under reduced pressure to yield a crude residue which was purified by flash chromatography on silica gel to give the title compound as a colorless oil (23 g, 65%). 1H NMR (300 MHz, CDCl3) δ 6.59 (s, 1H), 4.37 (q, 2H, J=7.2 Hz), 2.58 (t, 2H, J=7.2 Hz), 1.76-1.64 (m, 2H), 1.40 (t, 3H, J=6.6 Hz), 1.01 (t, 3H, J=7.2 Hz), 4.40 (q, 2H), 3.89 (s, 3H), 2.59 (t, 2H), 1.69 (2H), 1.37 (t, 3H), 1.01 (t, 3H); LC-MS: m/z=197 (MH)+.Step 42-Methyl-5-propyl-2H-pyrazole-3-carboxylic acid: 2-methyl-5-propyl-2H-pyrazole-3-carboxylic acid ethyl ester (29.4 g, 150 mmol) was suspended in 6N sodium hydroxide (120 mL, 720 mmol) and heated to 80° C. for 2 hours, cooled, diluted with water (100 mL) and acidified with 5N hydrochloric acid (200 mL) to give a precipitate which was filtered off and dried to give the title compound as a white solid (24.2 g, 96%). 1H NMR (300 MHz, CDCl3) δ 6.76 (s, 1H), 4.17 (s, 3H), 2.63 (t, 2H, J=7.2 Hz), 1.70-1.68 (m, 2H), 0.98 (t, 3H, J=7.2 Hz); LC-MS: m/z=169 (M+H)+;Step 52-Methyl-4-nitro-5-propyl-2H-pyrazole-3-carboxylic acid: A solution of 2-methyl-5-propyl-2H-pyrazole-3-carboxylic acid (22 g, 131 mmol) in concentrated sulfuric acid (98%, 85 mL) was heated to 50° C. and treated with a mixture of fuming nitric acid (95%, 7.7 mL) and concentrated sulfuric acid (98%, 18 mL), while keeping the reaction temperature between 50 and 55° C. The reaction mixture was kept for 8 hours at 50° C., cooled to ambient temperature, and slowly added to cold water (600 mL, 4° C.), keeping the temperature below 25° C. The precipitate was collected by filtration, and dried below 80° C. to give the title compound as a white solid (25 g, 90%). 1H NMR (300 MHz, CDCl3) δ 4.25 (s, 3H), 2.92 (t, 2H, J=7.5 Hz), 1.77-1.70 (m, 2H), 1.03 (t, 3H, J=7.2 Hz); LC-MS: m/z=214 (M+H)+Step 62-Methyl-4-nitro-5-propyl-2H-pyrazole-3-carboxamide: To a suspension of 2-methyl-4-nitro-5-propyl-2H-pyrazole-3-carboxylic acid (17.0 g, 79.8 mmol) in dry toluene (85 mL) was added a catalytic quantity of dimethylformamide (0.6 mL). The mixture was heated to 50° C. and thionyl chloride (17.1 g, 143.7 mmol) was added over 30 minutes. The reaction was stirred and heated at 55-60° C. for 6 hours. The solvent was removed, dry toluene (80 mL) was added and the mixture was cooled to 20° C. and cold (5° C.) concentrated ammonium hydroxide (100 mL) was added. The precipitate was filtered, washed with water and dried to give the title compound as an off-white solid (14.8 g, 87%). LC-MS: m/z=213 (M+H)+, 235 (M+Na)+.Step 74-Amino-2-methyl-5-propyl-2H-pyrazole-3-carboxamide: To a suspension of 2-methyl-4-nitro-5-propyl-2H-pyrazole-3-carboxamide (14.7 g, 69.3 mmol) in ethyl acetate (130 mL), was added 10% palladium on carbon (3.3 g). The mixture was reacted at 50° C. and 4 atm hydrogen pressure overnight. The reaction mixture was cooled, and the catalyst was filtered off and washed with ethyl acetate and dried over anhydrous magnesium sulfate, filtered and concentrated under reduced pressure to give the title compound (13.8 g, 98%). 1H NMR (300 MHz, CDCl3) δ 4.12 (s, 3H), 2.84 (s, 2H), 2.55 (t, 2H, J=7.2 Hz), 1.71-1.61 (m, 2H), 0.99 (t, 3H, J=7.2 Hz); LC-MS: m/z=183 (MH)+Step 82-Methyl-4-(2-propoxybenzoylamino)-5-propyl-2H-pyrazole-3-carboxamide: A solution of 2-propoxybenzoic acid (13.7 g, 76.1 mmol) and thionyl chloride (36.2 g, 304.4 mmol) in dry dichloromethane (80 mL) was heated for 3 hours at reflux. The solvent and excess thionyl chloride were distilled off under reduced pressure. The residue was taken up in dry dichloromethane (60 mL) and reacted with a solution of 4-amino-2-methyl-5-propyl-2H-pyrazole-3-carboxamide (12.6 g, 69.2 mmol), dry triethylamine (7 g, 69.2 mmol) and 4-(N,N-dimethylamino)pyridine (84.5 mg, 0.7 mmol) in dry dichloromethane (200 mL) at 0° C. Stirring was maintained for 1 hour, and the reaction mixture was successively washed with water (150 mL), saturated aqueous sodium carbonate solution (200 mL) and saturated brine (200 mL). The organic layer was dried over anhydrous magnesium sulfate and filtered. The filtrate was concentrated to about 60 mL, and then hexane (150 mL) was added to give precipitate product as a white solid (22 g, 92%). 1H NMR (300 MHz, CDCl3) δ 9.47 (s, 1H), 8.28 (d, 1H, J=7.8 Hz), 7.87 (br.s, 1H), 7.57-7.52 (m, 1H), 7.16-7.05 (m, 2H), 5.53 (s, 1H), 4.20 (t, 2H, J=6.6 Hz), 4.09 (s, 3H), 2.54 (t, 2H, J=7.5 Hz), 1.97-1.85 (m, 2H), 1.69-1.26 (m, 2H), 1.07 (t, 3H, J=7.2 Hz), 0.95 (t, 3H, J=7.5 Hz). LC-MS: m/z=345 (M+H)+Step 93-(5-Carbamoyl-1-methyl-3-propyl-1H-pyrazol-4-ylcarbamoyl)-4-propxy-benzenesulfonyl chloride: 2-Methyl-4-(2-propoxybenzoylamino)-5-propyl-2H-pyrazole-3-carboxamide (20 g, 58.1 mmol) was added to chlorosulfonic acid (81.3 g, 698 mmol) at 0° C. and the reaction was warmed to ambient temperature and stirred for 2 hours. The reaction mixture was poured into ice water (800 g) and mechanically stirred for 1 hour to give a white solid, which was filtered and washed with water. Following standard extractive work up, the solvent was evaporated under reduced pressure to yield the title compound (8 g, 31%). 1H NMR (300 MHz, CDCl3) δ 9.19 (s, 1H), 8.97 (s, 1H), 8.19 (t, 1H, J=8.9 Hz), 7.56 (br. s, 1H), 4.35 (t, 2H, J=6.6 Hz), 4.07 (s, 3H), 2.53 (t, 2H, J=7.5 Hz), 2.06-1.94 (m, 2H), 1.78-1.60 (m, 2H), 1.18 (t, 3H, J=7.5 Hz), 0.95 (t, 3H, J=7.2 Hz); LC-MS: m/z=443.1 (M+H)+Step 102-Methyl-4-{5-[2-(1-methyl-pyrrolidin-2-yl)-ethylsulfamoyl]-2-propoxy-benzoylamino}-5-propyl-2H-pyrazole-3-carboxamide: To a solution of 3-(5-carbamoyl-1-methyl-3-propyl-1H-pyrazol-4-ylcarbamoyl)-4-propoxy-benzenesulfonyl chloride (2.12 g, 4.8 mmol) and dry triethylamine (0.5 g, 4.8 mmol) in dichloromethane (20 mL), was added 2-(2-aminoethyl)-1-methylpyrrolidine (0.6 g, 4.8 mmol) at 0° C. The reaction was warmed to ambient temperature, stirred for 1 hour at ambient temperature, and diluted with dichloromethane (40 mL). Following standard extractive work up, the solvent was evaporated under reduced pressure to yield the title compound (2.2 g) which was used directly in the next step. LC-MS: m/z=535 (M+H)+Step 113-(1-Methyl-7-oxo-3-propyl-6,7-dihydro-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-N-[2-(1-methyl-pyrrolidin-2-yl)-ethyl]-4-propoxy-benzenesulfonamide: Potassium tert-butoxide (0.9 g, 8.0 mmol) was added to a solution of crude 2-methyl-4-{5-[2-(1-methyl-pyrrolidin-2-yl)-ethylsulfamoyl]-2-propoxy-benzoylamino}-5-propyl-2H-pyrazole-3-carboxamide (2.14 g, 4.0 mmol) in dry tert-butanol (50 mL), and the mixture was heated to reflux for 8 hours. The reaction mixture was cooled to ambient temperature and diluted with ethyl acetate (300 mL). Following standard extractive work up, the solvent was evaporated under reduced pressure to yield a crude residue which was purified by flash chromatography to give the title compound (1.1 g, 53%).1H NMR (300 MHz, CDCl3) δ 10.90 (broad s, 1H), 8.93 (s, 1H), 7.96 (d, 1H, J=8.7 Hz), 7.15 (d, 1H, J=8.7 Hz), 4.28-4.24 (m, 3H), 4.24 (s, 2H), 3.13 (t, 3H, J=6.9 Hz), 2.93 (t, 3H, J=7.8 Hz), 2.56 (s, 1H), 2.40 (s, 3H), 2.26-2.24 (m, 1H), 2.10-1.99 (m, 2H), 1.89-1.80 (m, 4H), 1.67 (s, 3H, J=7.2 Hz), 1.56-1.52 (m, 1H), 1.22 (t, 3H, J=7.5 Hz), 1.03 (t, 3H, J=7.2 Hz);LC-MS: m/z=517 (MH)+.........................References
- Udenafil Information
- Zydena (udenafil) product information page. Dong-A Pharmaceutical. Retrieved on April 13, 2009.
- Udenafil: efficacy and tolerability in the management of erectile dysfunction.
- http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3607490/
- British Journal of Pharmacology, 2008 , vol. 153, 7 PG. 1568 - 1578
- Arzneimittel-Forschung/Drug Research, 2009 , vol. 59, 12 pg. 641 - 646
- Chemical and Pharmaceutical Bulletin, 2011 , vol. 59, 9 PG. 1083 - 1088
- WO2010/13925 A2, ...
- US2010/173915 A1
- WO2010/95849 A2,
- WO2007/114534 A1, .....
- Life Sciences, 2004 , vol. 75, 9 pg. 1075 - 1083 ..............mp 162 - 164 °C
- US2008/194529 A1,
WO2008100886A1 * Feb 12, 2008 Aug 21, 2008 Auspex Pharmaceuticals Inc Preparation and use of deuterated udenafil analogues as highly selective pde5 modulators for the treatment of erectile dysfunction US6333330 * Oct 22, 1999 Dec 25, 2001 Pfizer Inc. Pyrazolopyrimidinone CGMP PDE5 inhibitors for the treatment of sexual dysfunction US20040029891 * Sep 2, 2003 Feb 12, 2004 Pfizer Inc. Use of PDE5 inhibitors in the treatment of polycystic ovary syndrome WO1993006104A1 * Sep 4, 1992 Apr 1, 1993 Pfizer Pyrazolopyrimidinone antianginal agents WO1994028902A1 * May 13, 1994 Dec 22, 1994 Peter Ellis Pyrazolopyrimidinones for the treatment of impotence WO1996016657A1 * Oct 16, 1995 Jun 6, 1996 Simon Fraser Campbell Bicyclic heterocyclic compounds for the treatment of impotence WO1998049166A1 * Apr 10, 1998 Nov 5, 1998 Mark Edward Bunnage PYRAZOLOPYRIMIDINONES WHICH INHIBIT TYPE 5 CYCLIC GUANOSINE 3',5'-MONOPHOSPHATE PHOSPHODIESTERASE (cGMP PDE5) FOR THE TREATMENT OF SEXUAL DYSFUNCTION EP0463756A1 * Jun 7, 1991 Jan 2, 1992 Pfizer Limited Pyrazolopyrimidinone antianginal agents WO1993006104A1 * Sep 4, 1992 Apr 1, 1993 Pfizer Pyrazolopyrimidinone antianginal agents WO1998049166A1 * Apr 10, 1998 Nov 5, 1998 Mark Edward Bunnage PYRAZOLOPYRIMIDINONES WHICH INHIBIT TYPE 5 CYCLIC GUANOSINE 3',5'-MONOPHOSPHATE PHOSPHODIESTERASE (cGMP PDE5) FOR THE TREATMENT OF SEXUAL DYSFUNCTION WO2000027848A1 * Nov 10, 1999 May 18, 2000 Byoung Ok Ahn Pyrazolopyrimidinone derivatives for the treatment of impotence EP0463756A1 * Jun 7, 1991 Jan 2, 1992 Pfizer Limited Pyrazolopyrimidinone antianginal agents WO2004108726A1 May 14, 2004 Dec 16, 2004 Tianjin North Pharma Sci Tech 2-SUBSTITUTED PHENYL-5,7-DIALKYL-3,7-DIHYDROPYRROLE [2,3-d] PYRIMIDINE-4-ONE DERIVATIVES, THE PREPARATION AND THE PHARMACEUTICAL USE THEREOF US7741483 Mar 6, 2008 Jun 22, 2010 Yangtze River Pharmaceutical (Group) Co., Ltd. Process for making substituted pyrrolo[2,3-d]pyrimidine derivatives as inhibitors of phosphodiesterase 5

 ..........................................................................................3. GISADENAFIL
..........................................................................................3. GISADENAFIL
GISEDENAFIL
GISEDENAFIL BESYLATE
334826-98-1 free form
334827-98-4 (as besylate)- UK 369003
- UK-369,003
- UK0369,003
- UNII-S6G4R7DI1C
THERAPEUTIC CLAIM Treatment of lower urinary tract
symptoms associated with BPHCHEMICAL NAMES FREE FORM1. ........7H-Pyrazolo[4,3-d]pyrimidin-7-one, 5-[2-ethoxy-5-[(4-ethyl-1-
piperazinyl)sulfonyl]-3-pyridinyl]-3-ethyl-2,6-dihydro-2-(2-methoxyethyl)-2. .......5-{2-ethoxy-5-[(4-ethylpiperazin-1-yl)sulfonyl]pyridin-3-yl}-3-ethyl-2-(2-
methoxyethyl)-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one3.........1-(6-Ethoxy-5-[3-ethyl]-6,7-dihydro-2-(2-methoxyethyl)-7-oxo-2H-pyrazole[4,3-d]pyrimidin-5-yl]-3-pyridylsulfonyl)-4-ethylpiperazineMOLECULAR FORMULA C23H33N7O5SMOLECULAR WEIGHT 519.6CODE DESIGNATION UK-369,003CAS REGISTRY NUMBER 334826-98-15-[2-Ethoxy-5-(4-ethylpiperazin-1-ylsulfonyl)pyridin-3-yl]-3-ethyl-2-(2-methoxyethyl)-6,7-dihydro-2H-pyrazolo[4,3-d]pyrimidin-7-onePhosphodiesterase PDE5A Inhibitors , Treatment of Erectile DysfunctionPfizer (Originator)UK-369003 is a phosphodiesterase V (PDE V) inhibitor which had been under development for the treatment of erectile dysfunction, pulmonary hypertension and for the treatment of lower urinary tract symptoms, but no recent development has been reported for these indications. Trials for the treatment of benign prostatic hyperplasia were discontinued.Gisadenafil besylate (USAN)
D09622, 334827-98-4M.Wt:677.795-(2-ethoxy-5-(4-ethylpiperazin-1-ylsulfonyl)pyridin-3-yl)-3-ethyl-2-(2-methoxyethyl)-2H-pyrazolo[4,3-d]pyrimidin-7(6H)-one benzenesulfonate1-[[6-Ethoxy-5-[3-ethyl-4,7-dihydro-2-(2-methoxyethyl)-7-oxo-2H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-pyridinyl]sulfonyl]-4-ethylpiperazine MonobenzenesulfonateFormula:C23H33N7O5S.C6H6O3SCertificate of Analysis Download Biological Activity:Potent and selective PDE5 inhibitor (IC50: 1.23 nM) with improved selectivity over PDE6(PDE5/6 selectivity value 117 and >3000-fold selectivity over other PDEs).Gisadenafil has the potential for oral bioavailability and dose-proportional pharmacokinetics. Close analogue of Sildenafil (Viagra; Axon 2046) Gisadenafil besylate is a PDE5 inhibitor. Inhibition of PDE5 prevents the breakdown of cyclic phosphodiester secondary messenger molecules. This has the effect of prolonging and enhancing signal transduction.CLINICAL TRIALS...............................PAPERSBioorganic and Medicinal Chemistry, 2012 , vol. 20, 1 p. 498 - 509Scheme 1.Reagents and conditions: (i) 1-(3-dimethylaminopropyl)-3-ethyl-carbodiimide hydrochloride, hydroxybenzotriazole, di-isopropylethylamine, THF, 20 °C, 20 h; (ii) caesium carbonate, alkyl mesylate or alkyl chloride, DMF, 20 °C, 20 h; (iii) KHMDS, R1OH, 120 °C, 20 h.Scheme 2.Reagents and conditions: (i) KHMDS, nBuOH, 120–130 °C, pressure vessel (ii) TFA, CH2Cl2; (iii) methanesulphonyl chloride, NEt3, CH2Cl2; (iv) HOAc, NaCNBH3, CH2O (v) KHMDS, nBuOH, reflux.Scheme 3.Reagents and conditions: (i) caesium carbonate, RCl, DMF; (ii) 50 psi H2, 10% Pd/C (iii) 1-(3-dimethylaminopropyl)-3-ethyl-carbodiimide hydrochloride, HOBT, di-isopropylethylamine, THF, 20 °C, 20 h; (iv) KHMDS, ethanol, 120 °C, pressure vessel; (v) TFA, CH2Cl2; (vi) CH2O, HOAc, NaCNBH3; (vii) R1OH, KHMDS, 120 °C.Scheme 4.Reagents and conditions: (i) NaNO2, HCl, H2O; (ii) TFAA, Et2O; (iii) ethyl propynoate, xylene, reflux, 2 h; (iv) NaOH, H2O, dioxan; (v) HNO3/H2SO4, 40–55 °C; (vi) (COCl)2, CH2Cl2, DMF; (vii) NH3, THF; (viii) 10% Pd/C, EtOH, 60 psi H2, 20 °C, 14 h; (ix) acid chloride of 3, NEt3, CH2Cl2; (x) KHMDS, EtOH, 130 °C, 14 h, pressure vessel; (xi) methoxyethanol, KHMDS, reflux, 14 h..................................PAPERSOrg. Proc. Res. Dev., 2004, 8 (4), pp 674–679DOI: 10.1021/op0300241...............................PAPERSYousef Hajikarimian, Steve Yeo, Robert W. Ryan, Philip Levett, Christopher Stoneley and Paul SinghOrg Process Res Dev 2010, 14(4): pp 1027–1031Publication Date (Web): June 25, 2010 (Article)DOI: 10.1021/op100141gUK-369,003 was nominated for development as the lead candidate for treatment of benign prostatic hyperplasia (BPH). The free base was found to be moderately crystalline with a melting point of 168 °C. Solubility of the free base at physiological pH was found to be poor hence necessitating a comprehensive screen for a suitable salt form of the API. Benzenesulfonic acid was found to form the most suitable counterion for the API with a melting point of 248 °C and satisfied all our requirements for primary and secondary processing. The process for the formation of the benzenesulfonic acid salt involved the use of water/methyl ethyl ketone (4% water by volume) as the reaction medium. The water level at 4% ensured an optimum balance between product quality (purging of impurities) and the reaction yield. The cyclisation reaction (step 2/Scheme 01) involves the use of ethanol as the reaction media. Any residual amount of ethanol in the isolated step 2 product was therefore considered to be a considerable risk factor in the potential formation of ethyl besylate during the final step processing (step 3/Scheme 01).Scheme 1. Manufacturing route to UK-369,003-26aaCDI = carbonyl diimidazole; MEK = methyl ethyl ketone; EtOAc = ethyl acetate; KOtBu = potassium tertiary butoxide; EtOH = ethanol.........................SYNTHESISCompound 1E is also known as 5-[2-Ethoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2-[2-methoxyethyl]-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one, or alternatively as 1-{6-ethoxy-5-[3-ethyl-6,7-dihydro-2-(2-methoxyethyl)-7-oxo-2H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-pyridyl sulphonyl}-4-ethylpiperazine (the compound of Example 103 of WO 01/27113 and exemplified hereinafter as Example 1).Preparation 12,2-dimethoxybutane:Methyl ethyl ketone (672 mL) was charged to a 2 L round bottomed flask and stirred at room temperature before being treated with, trimethylorthoformate (763 mL) and para-toluenesulphonic acid (6.65 g, 0.5 mol %). Over a 15 min period the internal temperature rose to 46° C., so the reaction was cooled to 0° C. for 30 min. The reaction was then stirred at room temperature for 2 h. The reaction was then neutralised by pouring onto sodium carbonate (ca. 750 g) with constant stirring. The resultant slurry was filtered under vacuum and the resultant filtrate was distilled at atmospheric pressure. The fraction boiling in the range 118° C.-124° C. was collected as a colourless liquid, 582 g, 70%.1H NMR (CDCl3): δ=0.88 (3H, t), 1.24 (3H, s), 1.61 (2H, q), 3.17 (6H, s).Example 1 N-[3-Carbamoyl-5-ethyl-1-(2-methoxyethyl)-1H-pyrazol-4-yl]-2-ethoxy-5-(4-ethyl-1-piperazinyl sulfonyl) nicotinamide(a) Ethyl 3-ethyl-1H-pyrazole-5-carboxylate (IIA) from (IlI) and (V)To a stirred solution of 2,2-dimethoxybutane (10 g, 84.7 mMol) in CH2Cl2 (50 mL) under a nitrogen atmosphere at 0° C. was added pyridine (13.7 mL, 169.5 mMol). The reaction mixture was maintained at 0° C. and a solution of trichloroacetyl chloride (18.9 mL, 169.5 mMol) in CH2Cl2 (35 mL) was added over 1 hour with constant stirring. The yellow-orange solution begins to precipitate a white solid as the reaction progresses. The reaction mixture is allowed to warm to room temperature over 20 h. The reaction mixture was diluted with ethanol (150 mL) and re-cooled to 0° C. before treatment with hydrazine hydrate (8.2 mL, 169.5 mMol) as a solution in ethanol (35 mL) over 30 min. The reaction was heated to 50° C. and solvent was distilled at atmospheric pressure. The temperature was increased until the head temperature reached 78° C. Reflux was maintained for a further 2 h, before cooling to room temperature. The reaction mixture was diluted with water (250 mL) and ethanol was removed by evaporation at reduced pressure. The resultant mixture was extracted with CH2Cl2 (3×200 mL). The combined organics were dried (MgSO4), filtered and evaporated at reduced pressure to afford the title compound as a brown oil, 12.05 g, 85%.1H NMR (300 MHz, CDCl3): δ=1.20 (3H, t), 1.28 (3H, t), 2.67 (2H, q), 4.29 (2H, q), 6.55 (1H, s), 12.56 (1H, s).LRMS m/z=167.1 [M-H]+, C8H12N2O2 requires 168.2.(b) Ethyl 3-ethyl-1H-pyrazole-5-carboxylic acid (IIA) from (IIA) via route 1Aqueous sodium hydroxide solution (10M; 100 ml, 1.0 mol) was added dropwise to a stirred suspension of the title compound of Example (a) (66.0 g, 0.39 mol) in methanol and the resulting solution heated under reflux for 4 hours. The cool reaction mixture was concentrated under reduced pressure to ca. 200 ml, diluted with water (200 ml) and this mixture washed with toluene (3×100 ml). The resulting aqueous phase was acidified with concentrated hydrochloric acid to pH 4 and the white precipitate collected and dried by suction to provide the title compound (34.1 g). δ (DMSOd6): 1.13 (3H,t), 2.56 (2H,q), 6.42 (1H,s).(c) 4-Nitro-3-n-propyl-1H-pyrazole-5-carboxylic acidFuming sulphuric acid (17.8 ml) was added dropwise to stirred, ice-cooled fuming nitric acid (16.0 ml), the resulting solution heated to 50° C., then 3-n-propyl-1H-pyrazole-5-carboxylic acid (Chem. Pharm. Bull., 1984, 32,1568; 16.4 g, 0.106 mol) added portionwise over 30 minutes whilst maintaining the reaction temperature below 60° C. The resulting solution was heated for 18 hours at 60° C., allowed to cool, then poured onto ice. The white precipitate was collected, washed with water and dried by suction to yield the title compound (15.4 g), m.p. 170-172° C. Found: C, 42.35; H, 4.56; N, 21.07. C7H9N3O4requires C, 42.21; H, 4.55; N, 21.10%. δ (DMSOd6): 0.90 (3H,t), 1.64 (2H,m), 2.83 (2H,m), 14.00 (1 H,s).(d) 3-Ethyl-4-nitro-1H-pyrazole-5-carboxylic acid (IIA) to (AA) via route 2Obtained from the title compound of Example (b), by analogy with the process of Example (c), as a brown solid (64%). δ (DMSOd6): 1.18 (3H,t), 2.84 (2H,m), 13.72 (1 H,s).(e) 4-Nitro-3-n-propyl-1H-pyrazole-5-carboxamideA solution of the title compound of Example (c) (15.4 g, 0.077 mol) in thionyl chloride (75 ml) was heated under reflux for 3 hours and then the cool reaction mixture evaporated under reduced pressure. The residue was azeotroped with tetrahydrofuran (2×50 ml) and subsequently suspended in tetrahydrofuran (50 ml), then the stirred suspension ice-cooled and treated with gaseous ammonia for 1 hour. Water (50 ml) was added and the resulting mixture evaporated under reduced pressure to give a solid which, after trituration with water and drying by suction, furnished the title compound (14.3 g).m.p. 197-199° C. Found: C, 42.35; H, 5.07; N, 28.38. C7H10N4O3 requires C, 42.42; H, 5.09; N, 28.27%. δ (DMSOd6): 0.90 (3H,t), 1.68 (2H,m), 2.86 (2H,t), 7.68 (1 H,s), 8.00 (1 H,s).(f) 3-Ethyl-4-nitro-1H-pyrazole-5-carboxamide BA from AA via route 3Obtained from the title compound of Example (d), by analogy with Example (e), as a white solid (90%). δ (DMSOd6): 1.17 (3H,t), 2.87 (2H,m), 7.40 (1H,s), 7.60 (1H,s), 7.90 (1H,s). LRMS: m/z 185 (M+l)+.(g)(i) 5-Ethyl-1-(2-methoxyethyl)-4-nitro-1H-pyrazole-3-carboxamide CA from BA via route 4A mixture of 3-ethyl-4-nitro-1H-pyrazole-5-carboxamide (2.5 kg, 13.6 Mol), sodium carbonate (1.8 Kg, 17.0 Mol) and 2-bromoethyl methyl ether (1.98 kg, 14.2 Mol) in THF (22.5 L) and water (2.5 L) was heated under reflux and stirred for 20 hours. The mixture was cooled to ambient temperature and CH2Cl2 (67.5 L) and water (22.5 L) were added. The resultant organic and aqueous layers were separated. The aqueous phase was extracted with CH2Cl2 (22.5 L) and the combined organic solution was distilled under atmospheric pressure and replaced with ethyl acetate (33 L) to a final volume of 17 L. The cooled mixture was granulated at ambient temperature for 2 hours, filtered and washed with ethyl acetate (2.5 L). This afforded 5-ethyl-1-(2-methoxyethyl)-4-nitro-1H-pyrazole-3-carboxamide as a white crystalline solid, 2.10 kg, 57%. m.p.=140° C. Found: C, 44.46; H, 5.79; N, 23.01. C9H14N4O4 requires C, 44.63; H, 5.79; N, 23.14%.δ (CDCl3): 1.18 (3H, t), 2.98 (2H, q), 3.22 (3H, s), 3.77 (2H, t), 4.28 (2H, q), 6.03 (1H, s), 7.36 (1H, s).LRMS: m/z=243 (M+1)+(g)(ii) 5-Ethyl-1-(2-methoxyethyl)-4-nitro-1H-pyrazole-3-carboxamide.A mixture of 3-ethyl-4-nitro-1H-pyrazole-5-carboxamide (25 g, 0.136 Mol), sodium carbonate (18 g, 0.17 Mol) and sodium iodide (20.4 g, 0.136 Mol) were suspended in ethyl methyl ketone (125 mL) at room temperature. 2-bromoethyl methyl ether (12.8 mL, 0.142 Mol) was added and the mixture was heated to reflux and stirred for 70 hours. The mixture was cooled to ambient temperature and water (250 mL) was added. The resultant slurry was warmed to reflux and held at that temperature for 30 min before cooling to room temperature. The resultant precipitate was granulated at room temperature for 3 h, filtered and vacuum dried to afford 5-ethyl-1-(2-methoxyethyl)-4-nitro-1H-pyrazole-3-carboxamide as a yellow crystalline solid 24.3 g, 74%. Data as reported for Example (g)(i).(h) 4-Amino-5-ethyl-1-(2-methoxyethyl)-1H-pyrazole-3-carboxamide (IA) from CA via route 5A mixture of 5-ethyl-1-(2-methoxyethyl)-4-nitro-1H-pyrazole-3-carboxamide (20 g, 82.6 mMol) and 5% Pd/C (1 g) in methanol (200 mL) was pressurised at 50psi/25° C. in a sealed vessel and stirred for 15 hours. At the end of the reaction the mixture was filtered through arbocel and the filter cake was washed with methanol. The methanolic solution was distilled at atmospheric pressure and replaced with ethyl acetate to a final volume of 100 mL. The cooled mixture was granulated at ambient temperature for 2 h filtered and washed with ethyl acetate (20 mL) to afford 4-amino-5-ethyl-1-(2-methoxyethyl)-1H-pyrazole-3-carboxamide as a white crystalline solid, 15 g, 88%. m.p.=131° C. Found: C, 50.75; H, 7.62; N, 26.38. C9H16N4O2 requires C, 50.94; H, 7.55; N, 26.42%. δ (CDCl3): 1.20 (3H, t), 2.63 (2H, q), 3.32 (3H, s), 3.74 (2H, t), 3.95 (2H, s), 4.15 (2H, t), 5.27 (1H, s), 6.59 (1H, s).LRMS: m/z=213 (M+1)+(i) N-[3-Carbamoyl-5-ethyl-1-(2-methoxyethyl)-1H-pyrazol-4-yl]-2-ethoxy-5-(4-ethyl-1-piperazinyl sulfonyl) nicotinamide.2-ethoxy-5-(4-ethyl-1-piperazinylsulfonyl)nicotinic acid (2.31 kg, 6.73 Mol) was suspended in ethyl acetate (16.2 L) and 1,1-carbonyldimidazole (1.09 kg, 6.73 Mol) was added at room temperature. The reaction mixture was heated at 45° C. for 40 minutes and then the reaction was stirred for a further 40 minutes at reflux. After cooling to ambient temperature 4-amino-5-ethyl-1-(2-methoxyethyl)-1H-pyrazole-3-carboxamide (1.5 kg, 7.06 Mol) was added to the cooled mixture, and the reaction stirred for a further 15 hours under reflux. The mixture was cooled filtered and the filter cake was washed with 90% water/10% ethyl acetate, (2 mL /g) to afford N-[3-carbamoyl-5-ethyl-1-(2-methoxyethyl)-1H-pyrazol-4-yl}-2-ethoxy-5-(4-ethyl-1-piperazinyl sulfonyl) nicotinamide as an off white crystalline solid, 3.16 kg, 88%. m.p.=156° C. Found: C, 51.33; H, 6.56; N, 18.36. C23H35N7O6S requires C, 51.40; H, 6.53; N, 18.25%.δ (CDCl3): 1.04 (3H, t), 1.22 (3H, t), 1.60 (3H, t), 2.44 (2H, q), 2.54 (4H, m), 2.96 (2H, q), 3.12 (4H, m), 3.36 (3H, s), 3.81 (2H, t), 4.27 (2H, t), 4.80(2H, q), 5.35(1H, s), 6.68 (1H, s), 8.66 (1H, d), 8.86 (1H, d), 10.51 (1H, s).LRMS: m/z=539 (M+1)+(i) 1-(6-Ethoxy-5-[3-ethyll-6,7-dihydro-2-(2-methoxyethyl)-7-oxo-2H-pyrazole[4,3-d]pyrimidin-5-yl]-3-pyridylsulfonyl)-4-ethylpiperazine•ethyl acetate solvate.GISADENAFILA mixture of N-[3-carbamoyl-5-ethyl-1-(2-methoxyethyl)-1H-pyrazol-4-yl}-2-ethoxy-5-(4-ethyl-1-piperazinyl sulfonyl) nicotinamide (1.18 kg, 2.2 Mol), potassium tert-butoxide (500 g, 4.4 moles) and ethyl acetate (193 g) in ethanol (11.8 L) was heated at 120° C. for 20 hours. The reaction mixture was then concentrated under reduced pressure, in total approx. 10 L of solvent were distilled. To the residue water (2.9 L) was added and the mixture stirred at room temperature while aqueous HCl was added until pH 7.5 was obtained. Ethyl acetate (7.5 L) was added and the two phase mixture was warmed to 55° C. The organic phase was separated and the aqueous phase was extracted with further ethyl acetate (3.0 L). The combined organic phases were distilled at atmospheric pressure to a final volume of 4 L. The precipitated solids were granulated at 5° C. for 1 h, filtered and washed with ethyl acetate (1.2 L) and dried under vacuum. This afforded 1-(6-Ethoxy-5-[3-ethyl]-6,7-dihydro-2-(2-methoxyethyl)-7-oxo-2H-pyrazole[4,3-d]pyrimidin-5-yl]-3-pyridylsulfonyl)-4-ethylpiperazine as a light yellow crystalline solid, 877 g, 78%. m.p.=157° C. Found: C, 52.65; H, 6.46; N, 17.76. C23H33N705S. 0.2 C2H5CO2CH3 requires C, 53.21; H, 6.49; N, 18.25%.δ (CDCl3): 1.07 (3H, t), 1.42 (3H, t), 1.61 (3H, t), 2.44 (2H, q), 2.57 (4H, m), 3.08 (2H, q), 3.15 (4H, m), 3.32 (3H, s), 3.92 (2H, q), 4.48 (2H, q), 4.77 (2H, q), 8.65 (1H, d), 9.06 (1H, d). The spectrum also has signals that correspond to a solvate with ethyl acetate.LRMS: m/z=520 (M+1)+.................Example 1021-(6-Ethoxy-5-f3-ethyll-6,7-dihvdro-2-(2-methoxyethvn-7-oxo-2r7-pyrazoler4.3- cf1pyrimidin-5-vn-3-pyridylsulfonyl)-4-ethylpiperazine»ethyl acetate solvate.To prepare the compound of Example 8 a mixture of Λ/-[3-carbamoyl-5-ethyl- 1 -(2-methoxyethyl)-1 /-/-pyrazol-4-yl}-2-ethoxy-5-(4-ethyl-1 -piperazinyl sulfonyl) nicotinamide (1.18 kg, 2.2 Mol), potassium tert-butoxide (500 g, 4.4 moles) and ethyl acetate (193 g) in ethanol (11.8 L) was heated at 120°C for 20 hours. The reaction mixture was then concentrated under reduced pressure, in total approx. 10 L of solvent were distilled. To the residue water (2.9 L) was added and the mixture stirred at room temperature while aqueous HCl was added until pH 7.5 was obtained. Ethyl acetate (7.5 L) was added and the two phase mixture was warmed to 55°C. The organic phase was separated and the aqueous phase was extracted with further ethyl acetate (3.0 L). The combined organic phases were distilled at atmospheric pressure to a final volume of 4L. The precipitated solids were granulated at 5°C for 1 h, filtered and washed with ethyl acetate (1.2 L) and dried under vacuum. This afforded 1 -(6-Ethoxy-5-[3-ethyl]-6,7-dihydro-2-(2-methoxyethyl)-7-oxo- 2H-pyrazole[4,3-o pyrimidin-5-yl]-3-pyridylsulfonyl)-4-ethylpiperazine as a light yellow crystalline solid, 877 g, 78%. m.p. = 157°C. Found: C, 52.65; H, 6.46; N, 17.76. C23H33N705S. 0.2 C2H5C02CH3 requires C, 53.21 ; H, 6.49; N, 18.25%.δ(CDCI3): 1.07 (3H, t), 1.42 (3H, t), 1.61 (3H, t), 2.44 (2H, q), 2.57 (4H, m), 3.08 (2H, q), 3.15 (4H, m), 3.32 (3H, s), 3.92 (2H, q), 4.48 (2H, q), 4.77 (2H, q), 8.65 (1 H, d), 9.06 (1 H, d). The spectrum also has signals that correspond to a solvate with ethyl acetate.LRMS: m/z = 520 (M+1)+Example 1031-(6-ethoxy-5-r3-ethyl-6.7-dihvdro-2-(2-methoxyethvn-7-oxo-2H-pyrazolor4.3- dlpyrimidin-5-vn-3-pyridylsulfonyl)-4-ethylpiperazineGISADENAFIL10g (0.019 mol) of the compound of Example 8 and Example 102, 1-{6- ethoxy-5-[3-ethyl-6,7-dihydro-2-(2-methoxyethyl)-7-oxo-2H-pyrazolo[4,3- d]pyrimidin-5-yl]-3-pyridylsulfonyl}-4-ethylpiperazine ethyl acetate solvate, was charged followed by 12ml/g (120mls) of 16% water in ethyl alcohol. The slurry was heated to reflux to yield a solution and 6ml/g (60mls) distilled off at atmospheric pressure. The solution was then cooled to room temperature with crystallisation occurring at 40°C. The slurry was then cooled to 5-10°C and granulated for 30 minutes following which it was filtered and washed with 2ml/g ethyl alcohol (20 mis). The damp solid was dried in vacuo overnight at 55-60 °C to yield a white crystalline solid. (Yield 7.6g, 76%). Melting Point 162- 165°C.δ (CDCI3): 1.05 (3H,t), 1.42 (3H,t), 1.58 (3H,t), 2.43 (2H,q), 2.57 (4H,t), 3.09 (2H, t), 3.15 (4H,t), 3.30 (3H,s), 3.93 (2H,t), 4.48 (2H,t), 4.90 (2H,q), 8.65 (1 H,d), 9.05 (1 H,d), 10.65 (1 H,s).In the process of Example 103, water and pharmaceutically acceptable alcohols such as methanol, ethanol, propanol, butanol and mixtures thereof can be used to prepare the compound of Examples 8 and 102.BESYLATE SALTExample 104 1-(6-ethoxy-5-r3-ethyl-6,7-dihvdro-2-(2-methoxyethyl)-7-oxo-2H-pyrazolor4.3- d]pyrimidin-5-yl]-3-pyridylsulfonyl)-4-ethylpiperazine benzene-sulfonate salt.170g (0.33 mol) of the compound of Example 103, 1-{6-ethoxy-5-[3-ethyl-6,7- dihydro-2-(2-methoxyethyl)-7-oxo-2H-pyrazolo[4,3- d]pyrimidin-5-yl]-3- pyridylsulfonyl}-4-ethylpiperazine, was charged followed by a water/ 2- butanone (4% v/v) at 10 ml/g (1.7 litres) and warmed to reflux. 53g (0.33 mol) of benzene sulfonic acid dissolved in water (23mls, resulting in 70 % w/w solution) was added to the refluxing solution over 30 minutes.5.3ml/g (0.9 litres) of 2-butanone were striped and replaced and the slurry cooled. The slurry was cooled to 5-10°C and granulated for 2 hours after which it was filtered and washed with 2ml/g (0.3 litres) of 2-butanone. The salt was dried overnight in vacuo at 55-60°C to yield a white crystalline solid. Yield 215g, 96.4%. Mpt 242-244°C. δ (DMSO): 1.17 (3H, t), 1.28 (3H, t), 1.35 (3H, t), 2.73 (2H, q), 2.97 (2H, q), 3.2 (3H, s), 3.58 (2H, t), 3.78 (3H, t), 3.81 (2H, t), 4.49 (2H, t) 4.51 (2H, q), 7.29-7.33 (3H, m), 7.57-7.60 (2H, m), 8.28 (1 H, d), 8.73 (1 H, d), 9.13 (1 H,s), 11.90(1 H,s).The powder X-ray diffraction (PXRD) pattern for this salt, having Mpt 242- 244°C, was determined using a Siemens D5000 powder X-ray diffractometer fitted with a theta-theta goniometer, automatic beam divergence slits, a secondary monochromator and a scintillation counter. The specimen was rotated whilst being irradiated with copper K-alpha1 X-rays (Wavelength = 1.5046 Angstroms) filtered with a graphite monochromator (λ = 0.15405nm) with the X-ray tube operated at 40 kV/mA. The main peaks (in degrees θ) of the PXRD pattern are illustrated in Table I.TableThe same besylate salt, as defined by the XRD pattern described in Table 1 , when made via alternative routes can have a melting point in the range of from 235-246°C (measured using a Perkin Elmer DSC7 at a heating rate of 20°C/minute).References1 The discovery of UK-369003, a novel PDE5 inhibitor with the potential for oral bioavailability and dose-proportional pharmacokinetics
Bioorg Med Chem 2012, 20(1): 498.............MP 161 - 162 °C2. Hajikarimian, Y.; Yeo, S.; Ryan, R.W.; Levett, P.; Stoneley, C.; Singh, P.
Investigation into the formation of the genotoxic impurity ethyl besylate in the final step manufacturing process of UK-369,003-26, a novel PDE5 inhibitor
Org Process Res Dev 2010, 14(4): 10273. Bentham; Dawson; Dunn; Papadopoulos; Taylor; Mitchell; Snowden; Taylor
Organic Process Research and Development, 2004 , vol. 8, 4 PG. 674 - 679 .............AS ENTRY B- Bloch, W., et al.: Prostate, 33, 1 (1997)
- • Glowienke, S., et al.: Mutat. Res., 581, 23 (1997)
- • Chapple, C., et al.: Eur. Urol., 54, 563 (1997)
- • Elder, D., et al.: J. Pharm. Pharmacol., 61, 269 (1997)
PATENTS1. WO 20100623662. WO 20070721563 WO 20070721564.US2002/22732 A1,5.US2002/28799 A1,6.WO1998049166A1 * Apr 10, 1998 Nov 5, 1998 Mark Edward Bunnage PYRAZOLOPYRIMIDINONES WHICH INHIBIT TYPE 5 CYCLIC GUANOSINE 3',5'-MONOPHOSPHATE PHOSPHODIESTERASE (cGMP PDE5) FOR THE TREATMENT OF SEXUAL DYSFUNCTION WO1999054333A1 * Mar 25, 1999 Oct 28, 1999 Mark Edward Bunnage Pyrazolopyrimidinone cgmp pde5 inhibitors for the treatment of sexual dysfunction US4666921 * 15 окт 1985 19 май 1987 Ludwig Heumann & Co. Gmbh Pyrazole derivatives, processes for their preparation and pharmaceutical preparations containing these compounds US5808092 * 15 окт 1997 15 сен 1998 Ube Industries, Ltd. Process for preparing-1-ethyl-5-hydroxypyrazole US6015911 * 24 мар 1998 18 янв 2000 Dow Agrosciences Llc Process for preparing 1-alkyl-4-(2-chloro-3-alkoxy-4-alkylsulfonylbenzoyl)-5-hydroxypyrazole and related compounds EP0463756A1 7 июн 1991 2 янв 1992 Pfizer Limited Pyrazolopyrimidinone antianginal agents EP0812845A1 4 июн 1997 17 дек 1997 Pfizer Limited Process for preparing sildenafil EP0994115A2 11 окт 1999 19 апр 2000 Pfizer Limited Process for preparation of pyrazolo-(4,3-d)pyrimidin-7-ones and intermediates thereof EP0995750A1 15 окт 1999 26 апр 2000 Pfizer Inc. Pyrazolopyrimidinone cGMP PDE5 inhibitors for the treatment of sexual dysfunction WO1998049166A1 10 апр 1998 5 ноя 1998 Mark Edward Bunnage PYRAZOLOPYRIMIDINONES WHICH INHIBIT TYPE 5 CYCLIC GUANOSINE 3',5'-MONOPHOSPHATE PHOSPHODIESTERASE (cGMP PDE5) FOR THE TREATMENT OF SEXUAL DYSFUNCTION WO1999054333A1 25 мар 1999 28 окт 1999 Mark Edward Bunnage Pyrazolopyrimidinone cgmp pde5 inhibitors for the treatment of sexual dysfunction WO2001027112A1 4 окт 2000 19 апр 2001 Charlotte Moira Norfo Allerton 5-(2-substituted-5-heterocyclylsulphonylpyrid-3-yl)-dihydropyrazolo[4,3-d]pyrimidin-7-ones as phosphodiesterase inhibitors WO2001027113A2 11 окт 2000 19 апр 2001 Mark Edward Bunnage PYRAZOLO `4,3-d! PYRIMIDINE DERIVATIVES PDE5 inhibitors mirodenafil THANKS AND REGARD'SDR ANTHONY MELVIN CRASTO Ph.D GLENMARK SCIENTIST , NAVIMUMBAI, INDIAdid you feel happy, a head to toe paralysed man's soul in action for you round the clock need help, email or call meMOBILE-+91 9323115463web linkI was paralysed in dec2007, Posts dedicated to my family, my organisation Glenmark, Your readership keeps me going and brings smiles to my family
THANKS AND REGARD'SDR ANTHONY MELVIN CRASTO Ph.D GLENMARK SCIENTIST , NAVIMUMBAI, INDIAdid you feel happy, a head to toe paralysed man's soul in action for you round the clock need help, email or call meMOBILE-+91 9323115463web linkI was paralysed in dec2007, Posts dedicated to my family, my organisation Glenmark, Your readership keeps me going and brings smiles to my family
 -2012 April FDA-approved treatment for ED medication")


















































...............................
4. DASANTAFIL
 Dasantafil
Dasantafil
569351-91-3 CAS NO
405214-79-1 (racemate)
UNII-48P711MI2G, SCH 446132, D03657,
Molecular Formula: C22H28BrN5O5
Molecular Weight: 522.39222
Merck & Co. (Originator) IN PHASE 2
THERAPEUTIC CLAIM treatment of erectile dysfunction (phosphodiesterase (PDE) 5 isoenzyme inhibitor)
CHEMICAL NAMES
- 1H-purine-2,6-dione, 7-[(3-bromo-4-methoxyphenyl)methyl]-1-ethyl-3,7-dihydro-8-[[(1R,2R)-2-hydroxycyclopentyl]amino]-3-(2-hydroxyethyl)
- 7-(3-bromo-4-methoxybenzyl)-1-ethyl-8-[[(1R,2R)-2-hydroxycyclopentyl]amino]-3-(2-hydroxyethyl)-3,7-dihydro-1H-purine-2,6-dione
7-[(3-bromo-4-methoxyphenyl)methyl]-l-ethyl-8-[[(lR,2R)-2- hydroxycyclopentyl]amino]-3-(2-hydroxyethyl)purine-2,6-dione
Treatment of Erectile Dysfunction , Phosphodiesterase PDE5A Inhibitors
Dasantafil (SCH-446132) is a phosphodiesterase type 5 (PDE5) inhibitor which had been in early clinical development at Merck & Co. for the treatment of erectile dysfunction (ED); however, no recent development has been reported for this research. Phosphodiesterases regulate the tissue concentration of cyclic guanosine monophosphate (cGMP), which in turn triggers smooth muscle relaxation, allowing blood to flow into the penis and resulting in erection. PDE5 is the most abundant phosphodiesterase in the human corpus cavernosum, and as such its inhibition by dasantafil enhances erectile function by increasing the concentration of cGMP.
DASANTAFIL
PDE V inhibitor compounds and their use in treating a variety of physiological conditions are described in a number of patents {e.g., U.S. Pat. Nos. 5,409,934, 5,470,579, 5,939,419 and 5,393,755) and foreign publications (e.g., WO 93/23401 , WO 92/05176, WO 92/05175, and WO 99/24433).
Specific PDE V inhibitors have been found useful for specific indications. For example, the use of PDE V inhibitors for treating impotence has met with commercial success with the introduction of sildenafil citrate, vardenafil, and tadalafil (i.e., Viagra®, Levitra®, and Cialis®, respectively). The chemistry and use of Viagra®, including its mechanism of action in treating erectile dysfunction, are taught in EP 0 702 555 B1. Accordingly, it is an object of this invention to provide a method of using a PDE V inhibitor to treat a patient who has, or is at risk of, congestive heart failure, and/or other cardiovascular conditions.
Processes for preparing PDE V inhibitor compounds can be found in US
6,207,829, US 6,066,735, US 5,955,611 , US 5,939,419, US 5,393,755, US 5,409,934, US 5,470,579, US 5,250,534, WO 02/24698, WO 99/24433, WO 93/23401 , WO 92/05176, WO 92/05175, EP 740,668 and EP 702,555. One type of PDE V inhibitor compound contains a xanthine functionality in its structure. Xanthines can be prepared as described by Peter K. Bridson and Xiaodong Wang in 1 -Substituted Xanthines, Synthesis, 855 (July, 1995), which is incorporated herein by reference in its entirety. WO 02/24698, which is incorporated herein by reference in its entirety, teaches a class of xanthine PDE V inhibitor compounds useful for the treatment of impotence. A general process disclosed therein for preparing xanthine PDE V inhibitor compounds having the formula (I) follows:

(III) (I) (i) reacting a compound having the formula (III) with an alkyl halide in the presence of a base (introduction of R11 or a protected form of R11); (ii) (a) debenzylating and then (b) alkylating the compound resulting from step (i) with an alkyl halide, XCH2R1"; (iii) (a) deprotonating and then (b) halogenating the compound resulting from step (ii);
(iv) reacting the compound resulting from step (iii) with an amine having the formula RlvNH2; and (v) removing a protecting portion of Rn, if present, on the compound resulting from step (iv) to form the compound having the formula (I). R1, R", Rm and Rlv correspond to R1, R2, R3 and R4, respectively, in WO02/24698, and are defined therein. WO 02/24698 (pages 44 and 68-73) also teaches a synthesis for the following xanthine compound (identified therein as Compound 13 or Compound 114 of Table II): 1-ethyl-3,7-dihydro-8-[(1 R,2R)- (hydroxycyclopentyl) amino]-3-(2-hydroxyethyl)-7-[(3-bromo-4- methoxyphenyl)methyl]-1 H-purine-2,6-dione:

Compound 13. It would be beneficial to provide an improved process for preparing polycyclic xanthine PDE V inhibitor compounds
......................
Patent description
entry 129 is dasantafil
.....................
SYNTHESIS
14X' CHs ' B" tX is Experimental Procedure: Compound 114 in Table II (13)
1 (20.0 g, 74.0 mmol) was dissolved in dimethylformamide (370 mL) under nitrogen and (2-bromoethoxy)-terf-butyldimethylsilane (31.8 mL, 148 mmol) was added dropwise. The reaction was stirred at room temperature for 115 hrs., then diluted with ethyl acetate and washed with water several times.
The organic mixture was dried over potassium carbonate, filtered and concentrated under vacuum. Purification via flash chromatography (30/70 ethyl acetate/hexanes) yielded 2 (28.1 g, 88%).
1H NMR (400 MHz, CDCI3): δ 7.52 (s, 1 H), 7.29-7.39 (m, 5H), 5.49 (s,
2H), 4.25 (t, 2H, J = 6.0 Hz), 4.07 (q, 2H, J = 7.2 Hz), 3.93 (t, 2H, J =
6.0 Hz), 1.24 (t, 3H, J = 7.2 Hz), 0.75 (s, 9H), 0.08 (s, 6H). HRMS: Calcd for C22H32N403Si (M+H): 429.2322. Found: 429.2329.
To a solution of 2 (2.10 g, 4.89 mmol) in methanol (375 mL) was added ammonium formate (4.64g, 73.6 mmol) and 20% palladium hydroxide on carbon (980 mg). The reaction was heated to reflux for 1.5 hrs., then cooled to room temperature, filtered and concentrated under vacuum. Purification via flash chromatography (50/50 ethyl acetate/hexanes) yielded 3 (1.26 g, 94%).
1H NMR (400 MHz, CDCI3): δ 7.82 (s, 1 H), 4.33 (t, 2H, J = 6.0 Hz), 4.16
(q, 2H, J = 7.2 Hz), 3.99 (t, 2H, J = 6.0 Hz), 1.29 (t, 3H, J = 7.2 Hz),
0.78 (s, 9H), 0.06 (s, 6H). HRMS: Calcd for Cι5H26N4O3Si (M+H): 339.1852. Found: 339.1864. To 3 (970 mg, 2.86 mmol) was added dimethylformamide (25 mL), 3- bromo-4-methoxybenzyl bromide 15 (1.62 g, 5.79 mmol), and potassium carbonate (800 mg, 5.79 mmol) under nitrogen. The reaction mixture was stirred at room temperature for 21 hrs., then diluted with ethyl acetate and washed with water several times. The organic mixture was dried over potassium carbonate, filtered and concentrated under vacuum. Purification by flash chromatography (30/70 ethyl acetate/hexanes) yielded 10 (1.55 g, 100%).
1H NMR (400 MHz, CDCI3): δ 7.52 (s, 1 H), 7.51 (d, 1 H, J = 2.4 Hz),
7.30 (dd 1 H, J = 2.0 Hz, J = 8.4 Hz), 6.87 (d, 1 H, J = 8.8 Hz), 5.40 (s,
2H), 4.25 (t, 2H, J = 6.0 Hz), 4.07 (q, 2H, J = 7.0 Hz), 3.93 (t, 2H, J =
6.0 Hz), 3.88 (s, 3H), 1.25 (t, 3H, J = 7.0 Hz), 0.75 (s, 9H), 0.08 (s, 6H).
HRMS: Calcd for C23H33BrN4O4Si (M+H): 537.1533. Found: 537.1540.
To solution of 10 (1.50 g, 2.80 mmol) in tetrahydrofuran (24 mL) under nitrogen at -78 °C (dry ice/acetone bath) was added lithium diisopropylamide (2M in THF/heptane, 2.2 mL, 4.33 mmol). After stirring for thirty minutes, 1 ,2- dibromotetrafluoroethane (0.69 mL, 5.77 mmol) was added dropwise over five minutes. The reaction was stirred for 1.25 hrs. at -78 °C then quenched with saturated aqueous sodium bicarbonate and warmed to room temperature.
The mixture was extracted with dichloromethane, dried over potassium carbonate, filtered and concentrated under vacuum. Purification via flash chromatography (30/70 ethyl acetate/hexanes) yielded 11 (600 mg, 34%). 1H NMR (400 MHz, CDCI3): δ 7.60 (d, 1 H, J = 2.4 Hz), 7.35 (dd, 1 H, J =
2.0 Hz, J = 8.4 Hz), 6.84 (d, 1 H, J = 8.4 Hz), 5.45 (s, 2H), 4.21 (t, 2H, J = 5.6 Hz), 4.07 (q, 2H, J = 6.8 Hz), 3.90 (t, 2H, J = 5.6 Hz), 3.87 (s, 3H), 1.24 (t, 3H, J = 6.8 Hz), 0.73 (s, 9H), 0.08 (s, 6H). HRMS: Calcd for C23H32Br2N4O4Si (M+H): 615.0638. Found: 615.0633.
To 11 (1.89 g, 3.07 mmol) was added the amino alcohol hydrochloride salt (1.31 g, 12.27 mmol), diisopropylethylamine (15.4 mL), and 1-methyl-2- pyrrolidinone (15.4 mL). The reaction mixture was heated to 160 °C in a sealed tube for 13 hrs., then cooled to room temperature. Water was added, then the mixture was extracted with ethyl acetate and washed with water several times. The organic mixture was dried over potassium carbonate, filtered and concentrated under vacuum. Purification via flash chromatography (3/97 methanol/dichloromethane) yielded 12 (1.77 g, 90%).
1H NMR (400 MHz, CDCI3): δ 7.45 (d, 1 H, J = 2.0 Hz), 7.17 (dd, 1 H, J =
2.4 Hz, J = 8.6 Hz), 6.86 (d, 1 H, J = 8.4 Hz), 5.18-4.34 (m, 3H), 4.00- 4.23 (m, 5H), 3.86-3.98 (m, 6H), 3.69-3.79 (m, 1 H), 2.10-2.21 (m, 1 H), 1.99-2.10 (m, 1 H), 1.60-1.84 (m, 3H), 1.32-1.43 (m, 1 H), 1.24 (t, 3H, J = 7.2 Hz), 0.75 (s, 9H), 0.07 (d, 6H, J = 4.0 Hz). HRMS: Calcd for C28H43BrN5θ5Si (M+H): 636.2217. Found: 636.2207.
12 (1.77 g, 2.78 mmol) was dissolved in tetrahydrofuran (28 mL) under nitrogen and tetrabutylammonium fluoride (1M in THF, 28 mL) was added dropwise. The reaction was stirred at room temperature for 15 hrs., then diluted with dichloromethane and washed with water several times. The organic mixture was dried over potassium carbonate, filtered and concentrated under vacuum. Purification via flash chromatography (3/97 methanol/dichloromethane) yielded 13 (compound no. 114 in Table II) (760 mg, 52%).
DASANTAFIL
1H NMR (400 MHz, CDCI3):
δ 7.47 (d, 1 H, J = 2.0 Hz), 7.19 (dd, 1 H, J =2.0 Hz, J = 8.4 Hz), 6.88 (d, 1 H, J = 8.4 Hz), 5.25 (s, 2H), 5.09 (s, 1H), 4.21-4.27 (m, 3H), 4.06 (q, 2H, J = 7.0 Hz), 3.90-3.97 (m, 3H), 3.89 (s, 1 H), 3.74-3.82 (m, 1 H), 3.08 (s, 1 H), 2.12-2.22 (m, 1 H), 1.98-2.08 (m, 1 H), 1.60-1.86 (m, 3H), 1.33-1.43 (m, 1 H), 1.25 (t, 3H, J = 7.0 Hz),1.06-1.22 (m, 3H). HRMS: Calcd for C22H28BrN5O5 (M+H): 522.1352. Found: 522.1346.
2-Bromo-4-methyl anisole 14 (2.2 mL, 14.9 mmol) was dissolved in dichlomethane (30 mL) and N-bromosuccinimide (3.75 g, 16.4 mmol) was added followed by AIBN (26.0 mg). The reaction was heated to reflux for 19 hrs., then cooled to room temperature and the precipitate was filtered off. The filtrate was diluted with dichloromethane and washed with 0.5 M aqueous sodium bicarbonate, followed by water. The organic mixture was dried over sodium sulfate, filtered and concentrated under vacuum to yield 15 (4.16 g,
100%). The benzyl bromide was used as the crude material without further purification.
1H NMR (400 MHz, CDCI3): δ 7.59 (d, 1 H, J = 2.0 Hz), 7.30 (dd, 1 H, J =
2.4 Hz, J = 8.4 Hz), 6.85 (d, 1 H, J = 8.4 Hz), 4.37 (s, 2H), 3.90 (s, 3H).
General Synthesis of Compound No. 114 in Table II (13) a) Reacting 1 with an alkyl halide and base to form 2; b) Debenzylation of 2 to form 3; c) Alkylation of 3 with a benzyl halide to form 10; d) Deprotonation of 10 followed. by addition of a brominating agent to form 11 ; e) Displacement of bromo 11 with an amine to form 12; and f) Silyl ether cleavage of 12 to form compound no. 114 in Table II (13).
114 IN TABLE II./(13)
...............
GENERAL SCHEME
SYNTHESIOS
1A
9A 13A DASANTAFIL
SYNTHESIS
Compound 1A:
glycine-A/-r(4-methoxyphenyl)methyl1 ethyl ester
To a mixture of glycine ethyl ester hydrochloride (about 1.4 equiv) and potassium carbonate (about 1.0 equiv) was added anhydrous ethanol. The mixture
was stirred at about 40-45 °C for about 3 hours. Then, p-anisaldehyde (about 1.0
equiv.) was added, and the reaction mixture was stirred for a minimum of about 3 hours to provide an imine (not shown). Upon reaction completion (about <5.0 % p- anisaldehyde remaining by GC analysis), the reaction mixture was cooled to about 0-
10 °C. Then, an aqueous solution of sodium borohydride (about 0.50 equiv) was
added to the reaction mixture at a temperature of between about 0 °C and about 20
°C, and stirred for about 1 hour to provide Compound 1 A. Upon completion of the
reduction reaction, the reaction mixture was quenched with the slow addition of an aqueous solution of aqueous glacial acetic acid. After quenching, the reaction mixture was warmed to room temperature and filtered to remove solids. The filtrate was then concentrated under vacuum, followed by the addition of toluene and water to facilitate layer separation. Aqueous potassium carbonate solution was added to adjust the pH of the mixture to about 8-9. The organic layer was separated and the aqueous layer was extracted with toluene. The combined toluene extracts were concentrated to provide the product in about a 80-85% yield (based on GC and HPLC in solution assay). 1H NMR 400 MHz (CDCI3): δ 7.23 (d, J = 8.5 Hz, 2H), 6.85 (d, J = 8.5 Hz, 2H),
4.17 (q, J = 7.1 Hz, 2H), 3.78 (s, 3H), 3.73 (s, 2H), 3.38 (s, 2H), 1.88 (s, br, 1 H), 1.26
(t, J = 7.1 Hz, 3H); 13C NMR 100 MHz (CDCI3): δ 172.8, 159.2, 132.0, 129.9, 114.2,
61.1, 55.6, 53.1 , 50.4, 14.6.
Compound 2:
/V-cvanomethanimidic acid ethyl ester
To cyanamide (about 1.2 mole) was added triethylorthoformate (about 1.33 mole), and the reaction mixture was heated to about 85-95 °C for approximately 2 hours to form Compound 2. Estimated in-solution yield was about 95-100%. The product was optionally purified by vacuum distillation.
1H NMR 400 MHz (CDCI3): δ 8.38 (s, 1H), 4.28 (t, J = 6.7 Hz, 2H), 1.29 (t, J =
6.8 Hz, 3H); 13C NMR 100 MHz (CDCI3): δ 171.5, 113.4, 65.5, 13.1.
Compound 3A:
cis- and frans-glvcine Λ/-r(cvanoimino,methyl1-Λ/-r(4- methoxyphenvDmethvπ ethyl ester
A solution of Compound 1A (about 1.0 mole) in toluene was concentrated under vacuum to distill off toluene. Anhydrous tetrahydrofuran ("THF") was added to the concentrate, then Compound 2 (about 1.2 moles, obtained above) was added to that, and the solution was heated at reflux for about 1 hour. At this stage, the formation of Compound 3A was complete. Estimated in-solution yield was about
95% (about 2:1 mixture of cis and trans isomers). Compound 4A: 1H-imidazole-5-carboxylic acid, 4-amino-1-[(4- methoxyphenvDmethvn ethyl ester
Compound 3A (obtained above) was concentrated by distilling off THF. Then, anhydrous ethanol was added to afford a reaction mixture solution. Separately, potassium t-butoxide (about 0.15 mole) was dissolved in anhydrous ethanol to afford a solution. The potassium t-butoxide solution was added to the reaction mixture solution and heated to about 75-85 °C for about 1 hour. The overall in-solution yield of Compound 4A was about 85-90%.
1H NMR 400 MHz (CDCI3): δ 7.16 (s, 1H), 7.08 (d, J = 8.6 Hz, 2H), 6.82 (d, J
=8.7 Hz, 2H), 5.23 (s, 2H), 4.93 (s, br, 2H), 4.23 (q, J = 7.1 , 2H), 3.76 (s, 3H), 1.26 (t,
J = 7.1 Hz, 3H); 13C NMR 400 MHz (CDCI3): δ 160.9, 159.2, 139.0, 128.6, 128.5,
114.0, 101.8, 59.5, 55.2, 50.1 , 14.4.
Compound 5AK:
4A 5AK
1 -ethyl-3,7-dihydro-7-F(4-methoxyphenyl)methvπ-1 H-Purine-2.6- dione potassium salt
The reaction mixture containing Compound 4A in ethanol (obtained above) was added to diglyme and distilled under vacuum to remove the ethanol. After being cooled to room temperature, Λ/-ethylurethane (about 1.2 equiv.) was added and the
reaction mixture was heated to about 110-120 °C. A solution of potassium t-butoxide
(2.2 equiv.) in diglyme was added to the hot solution. The reaction mixture was cooled to room temperature. THF was added to precipitate additional product, which was filtered and washed to provide Compound Salt 5AK in 55-65% overall yield. The wet cake can be used as such for conversion to Compound 6A.
1H NMR (DMSO-de, 400 MHz): δ 7.73 (s, 1H) 7.31 (d, J = 8.6 Hz , 2H) 6.86 (d,
J = 8.6 Hz, 2H) 5.24 (s, 1 H) 3.88 (q, J = 6.8 Hz, 2H) 3.71 (s, 3H) 1.07 (t, J = 6.8 Hz, 3H); 13C NMR (DMSO-d6, 100 MHz): δ 161.1 , 159.0, 158.4, 157.2, 141.4, 131.0,
129.5, 114.1 , 105.6, 55.4, 48.2, 34.4, 14.3.
Optional Neutralization of Compound Salt 5AK to Compound 5A: Compound 5A: 1-ethyl-3,7-dihvdro-7-r(4-methoχyphenyl,methvπ-1 H-Purine-2,6- dione
The wet cake filtered solid of Compound Salt 5AK (obtained above) was suspended in water and then acidified to a pH of about 5 using glacial acetic acid. The resulting slurry was filtered to obtain the neutralized product, which was then washed with water and dried. The overall isolated yield of neutralized Compound 5A from Compound 1 A was about 45-55%. Spectroscopic data for neutralized Compound 5A was identical to that of Compound Salt 5AK.
Compound 6A:
3-r2-(acetyloxy,ethvn-1-ethyl-3,7-dihvdro-7-r(4- methoxyphenyl,methvπ-1H-purine-2,6-dione
To the wet cake filtered solid of Compound Salt 5AK (obtained above) were added tetrabutylammonium bromide (about 0.05 mole) and 2-bromoethyl acetate
(about 1.2 moles) in THF. After being heated to reflux for about 2 hours, part of the THF was distilled off, and isopropyl alcohol was added to the reaction mixture. The reaction mixture was then concentrated under reduced pressure and cooled to around room temperature. Water was added to precipitate the product. After being cooled to about 0-5 °C for about a few hours, the product was isolated by filtration. The wet cake was washed with aqueous isopropyl alcohol (about 30% in water), and dried under vacuum to afford Compound 6A as a pale yellow solid in about a 45- 55% overall yield (based on Compound 1A). The crude product may be purified further by decolorizing with Darco in methanol, followed by filtration and concentration to afford crystalline Compound 6A.
1H NMR (CDCI3 , 400 MHz): δ 7.54 (s, 1 H) 7.32 (d, J = 8.6 Hz, 2H) 6.90 (d, J =
8.6 Hz, 2H) 5.43 (s, 2H) 4.41 (m, 2H) 4.38 (m, 2H) 4.10 (q, J = 7.2 Hz, 2H) 3.79 (s,
3H) 1.96 (s, 3H) 1.25 (t, J = 7.2 Hz, 3H); 13C NMR (CDCI3 , 100 MHz): δ 171.1 ,
160.2, 155.3, 151.4, 148.9, 140.9, 130.1 , 127.7, 114.8, 107.5, 61.7, 55.6, 50.2, 42.4, 36.9, 21.2, 13.6.
After Optional Neutralization of Compound Salt 5AK to Compound 5A:
Compound 6A:
3-r2-(acetyloxy.ethvπ-1-ethyl-3,7-dihvdro-7-r.4- methoxyphenyl)methyn-1H-purine-2,6-dione
Acetonitrile was added to a mixture of Compound 5A (about 1.0 mole), anhydrous potassium carbonate (about 1.5 moles) and tetrabutylammonium hydrogen sulfate (about 0.05 mole). 2-bromoethyl acetate (about 1.5 moles) was added in three separate portions (0.72 mole in the beginning, another 0.45 mole after about 2 hours of reaction, and then the remaining 0.33 mole after about another
1 hour of reaction) during the course of the reaction at about 80-85 °C. The total reaction time was about 7 hours. The reaction mixture was cooled to about room temperature and filtered. The filtrate was concentrated. Aqueous isopropanol was added to crystallize the product. The product was filtered, washed with aqueous isopropanol, and dried to provide Compound 6A in about a 75-80% yield. Compound 7A: 8-bromo-1 -ethyl-3-r2-(acetyloxy)ethvπ-3,7-dihvdro-7-r(3-bromo-4- methoxyphenyl)methvπ-1 - -Purine-2,6-dione
Compound 6A (about 1 mole) and NBS (about 2.8 moles) were dissolved in
dry acetonitrile and agitated at about 15-20 °C. To this reaction mixture, a solution of
sulfuric acid (about 0.03 mol) in acetonitrile was added, while maintaining the
reaction temperature below about 25 °C. The reaction mixture was agitated at about
20-25 °C for about 12-15 hours until complete consumption of the starting material
was indicated. The reaction mixture was cooled to about 0-5 °C and a cold (about 5-
10 °C) aqueous solution of sodium sulfite was added, keeping the temperature below
about 10 °C. The reaction was agitated for about 2 hours at about 0-10 °C, and then
filtered. The isolated cake was washed with water, followed by methanol, then dried under a vacuum to obtain Compound 7A in about an 85% yield.
1H NMR (CDCIs, 400 MHz): D 7.60 (d, J=2.0 Hz, 1H), 7.35 (dd, J=8.4 Hz, 2.0 Hz, 1 H), 6.83 (d, J=8.4 Hz, 1 H), 5.43 (s, 2H), 4.35 (m, 4H), 4.05 (q, J=7.0 Hz, 2H), 3.85 (s, 3H), 1.96 (s, 3H), 1.23 (t, J=7.0 Hz, 3H); 13C NMR (CDCI3, 100 MHz): D 171.0, 156.2, 154.2, 150.8, 148.2, 138.3, 128.9, 128.7, 127.5, 112.1 , 112.0, 109.1 , 61.5, 56.5, 49.3, 42.5, 37.0, 21.0, 13.3. MS (ES) m/e 545.2 (M+H)+.
Compound 13A:
1-ethyl-3.7-dihvdro-8-r(1f?,2 )-(hvdroxycvclopentyl)amino1-3-(2- hvdroxyethvπ-7-r(3-bromo-4-methoxyphenvhmethvπ-1/--purine-2.6-dione
Compound 7A (about 1 mole) was combined with (R,R)-2-amino-1- cyclopentanol hydrochloride (Compound 8A, about 1.2 moles) and sodium bicarbonate (about 3 moles). To this reaction mixture was added N,N- dimethylacetamide ("DMA"), and the reaction mixture was agitated at about 135-140 °C for about 15-17 hours until complete consumption of the starting material was
indicated.
Compound 9A is an intermediate that is formed, but not isolated, from the
reaction mixture. The reaction mixture was then cooled to about 45-50 °C, and
tetrabutylammonium hydroxide (about 0.05 moles of about a 40% solution in water) was charged therein, followed by methanol. The reaction mixture was refluxed at
about 80-85 °C for about 8-9 hours until complete deprotection of the acetate group
was indicated. The reaction mixture was cooled to about 40-45 °C and concentrated
under vacuum. The pH of the reaction mixture was adjusted to about 5-6 with dilute
acetic acid, and the reaction mixture was heated to about 55-65 °C, and seeded with
a small amount of Compound 13A. The reaction mixture was then cooled to about
30-35 °C over a period of about 2 hours, and water was added over a period of
about 1 hour. The reaction mixture was further cooled to about 0-5 °C over a period
of about 1 hour, and agitated at that temperature for about 4 hours. The Compound 13A product was isolated by filtration, washed with water and dried to provide about an 85-90% yield.
9A 13A DASANTAFIL
1H NMR (CDCI3, 400 MHz): D 7.47 (d, J=2.1 Hz, 1 H), 7.18 (dd, J=8.4 Hz, 2.0 Hz, 1 H), 6.87 (d, J=8.4 Hz, 1H), 5.23 (s, 2H), 5.01 (s, 1 H), 4.22 (m, 2H), 4.15 (m, 1H), 4.05 (q, J=7.0 Hz, 2H), 3.93 (m, 3H), 3.88 (s, 3H), 3.77 (m, 1H), 2.95 (m, 1H), 2.15 (m, 1H), 2.05 (m, 1 H), 1.60-1.80 (m, 4H), 1.35 (m, 1 H), 1.23 (t, J=7.0 Hz, 3H); 13C NMR (CDCI3, 100 MHz): D 156.2, 154.0, 153.5, 151.8, 148.3, 132.6, 129.1 , 127.9, 112.5, 103.2, 79.5, 77.8, 63.2, 61.3, 56.7, 46.5, 45.9, 36.8, 32.9, 31.5, 21.4, 13.8. MS (ES) m/e 523.4 (M+H)+. Micronization
INTERPRETATION
1H NMR (CDCI3, 400 MHz): DELTA
7.47 (d, J=2.1 Hz, 1 H), SANDWICHED AROM H BETWEEN BROMO AND -CH2-PY RING
7.18 (dd, J=8.4 Hz, 2.0 Hz, 1 H), AROM H ORTHO TO -CH2-PH RING AND PARA TO BROMO
6.87 (d, J=8.4 Hz, 1H), AROM H ORTHO TO O ATOM OF PH RING
5.23 (s, 2H), CH2 OF N-CH2-PH RING
5.01 (s, 1 H), OH OR NH 1H OUT OF 3 NOS
4.22 (m, 2H), OH OR NH 2H OUT OF 3 NOS
4.15 (m, 1H), --NCH2CH2OH 1H OUT OF 4 NOS
4.05 (q, J=7.0 Hz, 2H), CH2 OF NCH2 CH3
3.93 (m, 3H), ---NCH2CH2OH 3H OUT OF 4 NOS
3.88 (s, 3H), -OCH3
3.77 (m, 1H), OH-CH OF CYCLOPENTANE RING
2.95 (m, 1H),NH-CH OF CYCLOPENTANE RING
2.15 (m, 1H),
2.05 (m, 1 H), 1H ON CYCLOPENTANE RING
1.60-1.80 (m, 4H), 4H ON CYCLOPENTANE RING
1.35 (m, 1 H), 1 H PARA TO SUBS IN CYCLOPENTANE RING
1.23 (t, J=7.0 Hz, 3H) --NCH2 C
H3
.............................
DASANTAFIL
REFERENCES
1 WANG Y ET AL: "DESIGN AND SYNTHESIS OF XANTHINE ANALOGUES AS POTENT AND SELECTIVE PDE5 INHIBITORS" BIOORGANIC & MEDICINAL CHEMISTRY LETTERS, OXFORD, GB, vol. 12, no. 21, 2002, pages 3149-3152, XP009014973 ISSN: 0960-894X
2. Peter K. Bridson and Xiaodong Wang in 1 -Substituted Xanthines, Synthesis, 855 (July, 1995)
PATENTS
1. WO 2002024698..
2. WO 2003101992..
3.WO 2010062366
4. WO 2007002125
5. WO 2006104870
6. WO 2005051368
7. US 2004167137
8. WO 2003101991
9.WO2006104870A2 , Schering Corp Methods of treating benign prostatic hyperplasia or lower urinary track symptoms by using pde 5 inhibitors
| WO2005012303A1 * | Jul 29, 2004 | Feb 10, 2005 | Kevin B Alton | Metabolite of xanthine phosphodiesterase 5 inhibitor and derivatives thereof useful for treatment of erectile dysfunction |
| US7312223 | Jul 29, 2004 | Dec 25, 2007 | Schering Corporation | Metabolite of xanthine Phosphodiesterase 5 inhibitor and derivatives thereof useful for treatment of erectile dysfunction |
| WO2002024698A1 * | Sep 17, 2001 | Mar 28, 2002 | Schering Corp | Xanthine phosphodiesterase v inhibitors |
| WO2003020724A1 * | Aug 26, 2002 | Mar 13, 2003 | Schering Corp | Polycyclic guanine phosphodiesterase v inhibitors |
| WO2003042216A1 * | Nov 7, 2002 | May 22, 2003 | Schering Corp | Polycyclic guanine derivative phosphodiesterase v inhibitors |
| DE4411660A1 * | Apr 5, 1994 | Oct 12, 1995 | Hoechst Ag | Verwendung von Xanthinderivaten zur Reduktion der pathologischen Hyperreagibilität eosinophiler Granulozyten, neue Xanthinverbindungen und Verfahren zu deren Herstellung |
| EP0430025A2 * | Nov 20, 1990 | Jun 5, 1991 | Hokuriku Pharmaceutical Co., Ltd. | Xanthine compound, method for preparing thereof, and a pharmaceutical composition comprising the same |
| JP3072480A * | Title not available | |||
| JP4279586A * | Title not available | |||
| JP5001065A * | Title not available | |||
| US5728686 * | Oct 31, 1996 | Mar 17, 1998 | Hoechst Aktiengesellschaft | Alkylxanthine phosphonates and alkylxanthine phosphine oxides and their use as pharmaceuticals |
| US6214992 * | Jun 4, 1997 | Apr 10, 2001 | Hoechst Aktiengesellschaft | Use of theophylline derivatives for the treatment and prophylaxis of states of shock, novel xanthine compounds and processes for their preparation |
| WO2003057200A2 * | 13 jan 2003 | 17 juli 2003 | Richard David Carr | Compositions comprising inhibitors of dpp-iv and nep enzymes for the treatment of diabetes |
| WO2003101991A1 * | 30 mei 2003 | 11 dec 2003 | Schering Corp | Xanthine phosphodiesterase v inhibitor polymorphs |
| WO2006026395A1 * | 26 aug 2005 | 9 maart 2006 | Richard Dixon | Endothelin a receptor (eta) antagonists in combination with phosphodiesterase 5 inhibitors (pde5) and uses thereof |
| US20020169174 * | 28 aug 2001 | 14 nov 2002 | Samuel Chackalamannil | Xanthine phosphodiesterase V inhibitors |
| US20030060627 * | 25 jan 2002 | 27 maart 2003 | Jordi Gracia Ferrer | 8-Phenyl-6, 9-dihydro-[1,2,4] triazolo[3,4-i]purin-5-one derivatives |
| US20040063731 * | 14 jan 2002 | 1 april 2004 | Hans-Michael Eggenweiler | Pharmaceutical formulation comprising pyrazolo[4,3-d]pyrimidines and endothelin receptor antagonists or thienopyrimidines and endothelin receptor antagonists |
| US2011142956 | 6-17-2011 | USE OF A PDE 5 INHIBITOR FOR TREATING AND PREVENTING HYPOPIGMENTARY DISORDERS |
| US7786301 | 8-32-2010 | Process for preparing xanthine phosphodiesterase V inhibitors and precursors thereof |
| US2009149480 | 6-12-2009 | METHODS OF USING PDE V INHIBITORS FOR THE TREATMENT OF CONGESTIVE HEART FAILURE |
| US7531544 | 5-13-2009 | Xanthine Phosphodiesterase V Inhibitors |
| US2009105282 | 4-24-2009 | METHODS OF TREATING BENIGN PROSTATIC HYPERPLASIA OR LOWER URINARY TRACT SYMPTOMS BY USING PDE 5 INHIBITORS |
| US2009074869 | 3-20-2009 | PHARMACEUTICAL FORMULATIONS |
| US2008051408 | 2-29-2008 | Use of a Pde 5 Inhibitor for Treating and Preventing Hypopigmentary Disorders |
| US7312223 | 12-26-2007 | Metabolite of xanthine phosphodiesterase 5 inhibitor and derivatives thereof useful for treatment of erectile dysfunction |
| US7268141 | 9-12-2007 | Xanthine phosphodiesterase V inhibitors |
| US2007122355 | 5-32-2007 | RAPIDLY ABSORBING ORAL FORMULATIONS OF PDE 5 INHIBITORS |
| US7192962 | 3-21-2007 | Xanthine phosphodiesterase V inhibitor polymorphs |
| US2007037831 | 2-16-2007 | Methods of using PDE 5 inhibitors for the treatment of congestive heart failure |
| US2007031349 | 2-9-2007 | Rapidly absorbing oral formulations of PDE 5 inhibitors |
| US2007004745 | 1-5-2007 | Methods of treating benign prostatic hyperplasia or lower urinary tract symptoms by using PDE 5 inhibitors |
| US7074923 | 7-12-2006 | Process for preparing xanthine phosphodiesterase V inhibitors and precursors thereof |
| US2006040962 | 2-24-2006 | Pharmaceutical formulations |
| US2005182077 | 8-19-2005 | Xanthine phosphodiesterase V inhibitors |
| US2005075349 | 4-8-2005 | Xanthine phosphodiesterase V inhibitors |
| US6821978 | 11-24-2004 | Xanthine phosphodiesterase V inhibitors |
| US2004229885 | 11-19-2004 | Xanthine phosphodiesterase V inhibitors |

Mirodenafil, 米罗那非 标准品
SYNTHESIS WILL BE UPDATED SOON
SK-3530
UNII-504G362H0H
862189-96-6 DIHYDROCHLORIDE
862189-95-5 (free base)
862189-95-5 (free base)
5-Ethyl-3,5-dihydro-2-[5-([4-(2-hydroxyethyl)-1-piperazinyl]sulfonyl)-2-propoxyphenyl]-7-propyl-4H-pyrrolo[3,2-d]pyrimidin-4-one
5-ethyl-2-f-5-[4-(2-hydroxyethyl)piperazine-1-sulfonyl]-2-phenylg -7-propoxypropyl-3,5-dihydropyrrolo-[3,2-d]-pyrimidin-4-one
5-(5-(4-(3-hydroxypropyl)piperazinylsulfonyl)-2-n-propoxyphenyl)-1-ethyl-3-n-propyl-1,6-dihydro-7H-pyrrolo[4,3-d]pyrimidin-7-one
2-(5-(4-(3-hydroxypropyl)piperazin-1-ylsulfonyl)-2-n-propoxyphenyl)-5-ethyl-7-n-propyl-3,5-dihydro-4H-pyrrolo[3,2-d]pyrimidin-4-one;
Launched – 2007
In2Gen (Originator)
SK Chemicals (Originator)
SK Chemicals (Originator)
Treatment of
Treatment of Erectile Dysfunction , hypertention
Treatment of Erectile Dysfunction , hypertention
Mirodenafil belongs to a class of drugs called PDE5 inhibitors, which many other erectile dysfunction drugs such as sildenafil, tadalafil, andvardenafil also belong to. It was developed by SK Chemicals Life Science and is marketed under the trade name of Mvix tab which comes in different doses (50 mg, 100 mg).
Mirodenafil is also available under the name of Mvix S ODF 50 mg as an orally dissolving film (ODF) which dissolves on the tongue without water. It is the first licensed medicine for the treatment of erectile dysfunction as a dosage form of film.
Mirodenafil is a newly developed oral phosphodiesterase type 5 inhibitor, currently under investigation as a treatment for erectile dysfunction (ED).
MIRODENAFIL米罗那非 标准品
Mirodenafil hydrochloride is a high selective PDE5 inhibitor commercialized by SK Chemicals which had been in early clinical development for the treatment of erectile dysfunction (ED). Early clinical studies had also been ongoing for the treatment of hypertension in patients taking amlodipine; however, no recent development has been reported for this research. The development of compound started in 1998 jointly by SK Chemicals and a bio-venture In2Gen.
Several clinical trials were conducted,[1][2][3] but mirodenafil has not been approved for use in the United States by the U.S. Food and Drug Administration.

CLINICAL STUDIES
Mirodenafil dihydrochloride
| ||||||||||||
The introduction of oral phosphodiesterase type 5 inhibitor therapy in 1998 revolutionized the treatment of erectile dysfunction. Erectile dysfunction is the most common sexual problem in men. It often has a profound effect on intimate relationships and quality of life. The analysis of pharmaceuticals is an important part of the drug development process as well as for routine analysis and quality control of commercial formulations. Whereas the determination of sildenafil citrate, vardenafil and tadalafil are well documented by a variety of methods, there are few publications about the determination of udenafil, lodenafil carbonate, mirodenafil and avanafil. The paper presents a brief review of the action mechanism, adverse effects, pharmacokinetics and the most recent analytical methods that can determine drug concentration in biological matrices and pharmaceutical formulations of these four drugs.
European patent applications EP-A-0463756 and EP-A-0526004 disclose certain pyrazolo 4,3-dpyrimidin-7-ones as cGMP PDE inhibitors, useful in the treatment of cardiovascular disorders such as angina, hypertension and heart failure. International application WO 94/28902 discloses their use for the treatment of impotence. 0017The present inventors have recently disclosed a series of pyrazolo4,3-dpyrimidin-7-one derivatives as PDE V inhibitors (Appln. No. KR 98-60436 and KR 99-7580). Herein a new series of pyrrolo4,33,2d-pyrimidin-74-one derivatives are prepared as PDE V inhibitors
Korean Patent No. 358083 discloses pyrrolopyrimidinone derivatives having good inhibition activity against PDE-5, a method of its preparation thereof, an intermediate compound used to prepare the same and their use for prevention and treatment of erectile dysfunction, pulmonary arterial hypertension, chronic obstructive pulmonary disease, benign prostatic hypertrophy and lower urinary tract diseases.
Of the pyrrolopyrimidinone derivatives disclosed in Korean Patent No. 358083, 5-ethyl-2-{5-[4- (2-hydroxyethyl)piperazin-1-ylsulfonyl]-2-n-propoxyphenyl}-7-n-propyl-l-3,5-dihydro-4 H-pyrrolo[3,2-d]pyrimidin-4-one (hereinafter, “SK-3530″) represented by the following formula (1 ) is an excellent selective inhibitor PDE-5 over other PDEs and is under clinical trial for the treatment of erectile dysfunction after passing through the preclinical stage.

The dihydrochloride salt (2HCI) of SK-3530 has been under investigation through the preclinical and clinical stages.
The SK-3530 dihydrochloride salt has good solubility and can be easily stabilized for pharmaceutical preparation. But, it has the following drawbacks.
First, because the SK-3530 dihydrochloride salt is hygroscopic, it easily absorbs moisture from the atmosphere and becomes discolored when the moisture content is high. And, due to the hygroscopic property, an anhydrous solvent condition and a dry air condition have to be provided to obtain a stable product. Second, the SK-3530 dihydrochloride salt should be kept at a temperature lower than room temperature because it does not show enough stability at room temperature. In particular, the SK-3530 dihydrochloride salt is labile to heat or light, and thus any prolonged exposure to heat or light results in various impurities.
Third, the SK-3530 dihydrochloride salt could corrode the punch during tablet ting due to its somewhat corrosive properties. This is because the SK-3530 dihydrochloride salt is a simple amorphous salt rather than being a stable crystalline acid addition salt or hydrate form. Thus, one of the two hydrochloric acid groups with a relatively weak ionic bond character may leave the molecule under severe conditions. As aforementioned, the SK-3530 dihydrochloride salt may be endowed with a sufficient stability for pharmaceutical preparation. But, some additional techniques and costs are needed due to the deficiency in intrinsic physicochemical property and stability of the compound.
MIRODENAFIL米罗那非 标准品
…………………………
The invention relates to a series of pyrrolopyrimidinone derivatives of the formula (1):
R1 ETHYL
R2=H
R3= PROPYL
R4 = PROPYL
R5=R5=SO2NR6R7, NR6R7 is 4-(3-hydroxypropyl)piperazinyl) IS MIRODENAFIL
ANALOGOUS METHOD
BELOW IS CUT PASTE OF R1 METHYL ANALOGUE ……………..R1 =METHYL AND NOT ETHYL ….CAUTION
Example 39 Preparation of
5-(5-(4-(2-hydroxyethyl)piperazinylsulfonyl)-2-n-propoxyphenyl)-1-methyl-3-n-propyl-1,6-dihydro-7H-pyrrolo[4,3-d]pyrimidin-7-one hydrochloride (a compound of the formula (1) wherein R5=SO2NR6R7, R1=CH3, R2=H, R3=CH2CH2CH3, R4=CH2CH2CH3; NR6R7 is 4-(2-hydroxyethyl)piperazinyl)
The titled compound was prepared as described in Example 2 by using 5-(5-(4-(2-hydroxyethyl)piperazinylsulfonyl)-2-n-propoxyphenyl)-1-methyl-3-n-propyl-1,6-dihydro-7H-pyrrolo[4,3-d]pyrimidin-7-one in place of 5-(2-ethoxy-5-(4-methylpiperazinylsulfonyl)phenyl)-1-methyl-3-n-propyl-1,6-dihydro-7H-pyrrolo[4,3-d]pyrimidin-7-one.
yield: 99%
mp 66.5° C. dec;
IR (neat) 3332 (NH and OH), 1676 (C═O), 1166 (SO2) cm−1;
1H NMR (DMSO-d6) δ 0.92 (t, J=7.2 Hz, 3H, CH2CH2CH3), 0.96 (t, J=7.2 Hz, 3H, OCH2CH2CH3), 1.56-1.80 (m, 4H, 2 CH2CH2CH3), 2.59 (t, J=7.5 Hz, 2H, CH2CH2CH3), 2.91 (br t, J=11.7 Hz, 2H, 2 SO2NCHax), 3.12-3.27 (m, 4H, NCH2CH2 and 2 SO2NCHeq), 3.58 (br d, J=11.7 Hz, 2H, 2 +HNCHax), 3.68-3.85 (m, 4H, CH2CH2OH and 2 +HNCHeq), 4.00 (s, 3H, NCH3), 4.15 (t, J=6.3 Hz, 2H, OCH2CH2CH3), 4.66 (br s, 1H, OH), 7.28 (s, 1H, H-2), 7.44 (d, J=9.0 Hz, 1H, H-3′), 7.89 (dd, J=9.0 Hz, 2.4 Hz, 1H, H-4′), 8.01 (d, J=2.4 Hz, 1H, H-6′), 10.85 (br s, 1H, NH+), 12.01 (br s, 1H, NH).
Example 42 Preparation of
5-(5-(4-(3-hydroxypropyl)piperazinylsulfonyl)-2-n-propoxyphenyl)-1-methyl-3-n-propyl-1,6-dihydro-7H-pyrrolo[4,3-d]pyrimidin-7-one (a compound of the formula (1) wherein R5=SO2NR6R7, R1=CH3, R2=H, R3=CH2CH2CH3, R4=CH2CH2CH3; NR6R7 is 4-(3-hydroxypropyl)piperazinyl)
The titled compound was prepared as described in Example 1 by using 5-(5-chlorosulfonyl-2-n-propoxyphenyl)-1-methyl-3-n-propyl-1,6-dihydro-7H-pyrrolo[4,3-d]pyrimidin-7-one and 1-(3-hydroxypropyl)piperazine in place of 5-(5-chlorosulfonyl-2-ethoxyphenyl)-1-methyl-3-n-propyl-1,6-dihydro-7H-pyrrolo[4,3-d]pyrimidin-7-one and 1-methylpiperazine.
yield: 94%
mp 162.5° C. dec (EtOAc/hexanes);
IR (neat) 3484, 3302 (NH and OH), 1669 (C═O), 1170 (SO2) cm−1;
1H NMR (CDCl3/TMS) δ 1.00 (t, J=7.5 Hz, 3H, CH2CH2CH3), 1.20 (t, J=7.5 Hz, 3H, OCH2CH2CH3), 1.64-1.80 (m, 4H, CH2CH2CH2OH and CH2CH2CH3), 1.99-2.11 (m, 2H, OCH2CH2CH3), 2.58-2.64 (m, 6H, NCH2CH2 and 2 NCH2), 2.71 (t, J=7.5 Hz, 2H, CH2CH2CH3), 3.08 (br s, 4H, 2 SO2NCH2), 3.71 (t, J=5.4 Hz, 2H, CH2CH2OH), 4.08 (s, 3H, NCH3), 4.26 (t, J=6.3 Hz, 2H, OCH2CH2CH3), 4.28 (br s, 1H, OH), 6.88 (s, 1H, H-2), 7.14 (d, J=8.7 Hz, 1H, H-3′), 7.77 (dd, J=8.7 Hz, 2.7 Hz, 1H, H-4′), 8.87 (d, J=2.7 Hz, 1H, H-6′), 10.69 (br s, 1H, NH); MS (FAB) m/z 532 (MH+).
Example 43 Preparation of
5-(5-(4-(3-hydroxypropyl)piperazinylsulfonyl)-2-n-propoxyphenyl)-1-methyl-3-n-propyl-1,6-dihydro-7H-pyrrolo[4,3-d]pyrimidin-7-one hydrochloride (a compound of the formula (1) wherein R5=SO2NR6R7, R1=CH3, R2=H, R3=CH2CH2CH3, R4=CH2CH2CH3; NR6R7 is 4-(3-hydroxypropyl)piperazinyl)
The titled compound was prepared as described in Example 2 by using 5-(5-(4-(3-hydroxypropyl)piperazinylsulfonyl)-2-n-propoxyphenyl)-1-methyl-3-n-propyl-1,6-dihydro-7H-pyrrolo[4,3-d]pyrimidin-7-one in place of 5-(2-ethoxy-5-(4-methylpiperazinylsulfonyl)phenyl)-1-methyl-3-n-propyl-1,6-dihydro-7H-pyrrolo[4,3-d]pyrimidin-7-one.
yield: 99%
mp 62.5° C. dec;
IR (neat) 3347, 3321 (NH and OH), 1689 (C═O), 1168 (SO2) cm−1;
1H NMR (DMSO-d6) δ 0.93 (t, J=7.5 Hz, 3H, CH2CH2CH3), 0.96 (t, J=7.5 Hz, 3H, OCH2CH2CH3), 1.57-1.87 (m, 6H, CH2CH2CH2OH and 2 CH2CH2CH3), 2.59 (t, J=7.5 Hz, 2H, CH2CH2CH3), 2.89 (br t, J=11.7 Hz, 2H, 2 SO2NCHax), 3.01-3.19 (m, 4H, NCH2CH2 and 2 SO2NCHeq), 3.44 (t, J=6.0 Hz, 2H, CH2CH2OH), 3.52 (br d, J=11.7 Hz, 2H, 2 +HNCHax), 3.79 (br d, J=11.7 Hz, 2H, 2 +HNCHeq), 4.00 (s, 3H, NCH3), 4.15 (t, J=6.6 Hz, 2H, OCH2CH2CH3), 4.71 (br s, 1H, OH), 7.29 (s, 1H, H-2), 7.44 (d, J=8.7 Hz, 1H, H-3′), 7.89 (dd, J=8.7 Hz, 2.4 Hz, 1H, H-4′), 8.02 (d, J=2.4 Hz, 1H, H-6′), 11.13 (br s, 1H, NH+), 12.05 (br s, 1H, NH).
……………………………
Synthesis from patent and some construction by me
you can synthesize as follows, A CHEMIST CAN PICK THIS UP, this is not available clearly anywhere
Chlorosulfonation of the methyl salicylate with ClSO3H in SOCl2 affords the Methyl 3-Chlorosulfonyl-6-hydroxybenzoate described below
THESE INTERMEDIATES FROM PATENT MAY HELP YOU
methyl salicylateX=CL, R8=ME- Methyl 3-Chlorosulfonyl-6-hydroxybenzoate
Example 1 EP1362858A1
- To a cooled solution of SOCl2 (156 g, 1. 31 mol) and ClSO3H (460 g, 3.94 mol) at 0°C was added slowly methyl salicylate (200 g, 1.31 mol) for 30 minutes, and the mixture was stirred at room temperature for 20 hours. The reaction mixture was poured slowly into the ice (2 kg) and H2O (3 L) mixture, and the resulting white precipitates were collected by filtration. The filtered solid was washed with H2O (3 L), air-dried for 2 days and then dried under vacuum at 40°C for 2 days to afford the titled product (232 g, 93%) as a white solid.
mp 76.5-77.5 °C (toluene/hexanes);
IR (neat) 1699 (C=O) cm-1;
1H NMR (CDCl3/TMS) δ 3. 90 (s, 3 H, OCH3), 6. 93 (d, J= 8. 7 Hz, 1 H, H-3), 7. 70 (dd, J= 8. 7 Hz, 2. 4 Hz, 1 H, H-4), 8. 03 (d, J= 2. 4 Hz, 1 H, H-6).
- Methyl 3-Chlorosulfonyl-6-hydroxybenzoate
Example 2 EP1362858A1
1-(2-hydroxyethyl)piperazine
R8=ME, W=N, n=2
- Methyl 2-Hydroxy-5-[4-(2-hydroxyethyl)piperazin-1-ylsulfonyl]benzoate
- To a mixture of 1-(2-hydroxyethyl)piperazine (27 mg, 0. 21 mmol) and K2CO3 (33 mg, 0. 24 mmol) in DMF (5 mL) was added methyl 3-chlorosulfonyl-6-hydroxybenzoate (50 mg, 0. 20 mmol), and the mixture was stirred at room temperature for 1 hour. The reaction mixture was washed with H2O (10 mL), and the aqueous layer was further extracted with 5% MeOH in CH2Cl2 (20 mL). The combined organic layer was dried (MgSO4), filtered, and the filtrate was evaporated to dryness under reduced pressure. The crude residue was purified by MPLC on silica gel (5% MeOH in CH2Cl2) to afford the titled compound (59 mg, 86%) as white solid.
mp 152 °C (dec) (CH2Cl2/ether);
IR (neat) 1685 (C=O) cm-1;
1H NMR (CDCl3/TMS) δ 2. 30 (br s, 1 H, CH2OH), 2. 63 (t, J = 5. 4 Hz, 2 H, NCH 2CH2O), 2. 70 (m, 4 H, 2 NCH2), 3. 12 (m, 4 H, 2 SO2NCH2), 3. 64 (t, J= 5. 4 Hz, 2 H, NCH2CH 2O), 4. 01 (s, 3 H, OCH3), 7. 12 (d, J= 8. 7 Hz, 1 H, H-3), 7. 81 (dd, J= 8. 7 Hz, 2. 4 Hz, 1 H, H-4), 8. 26 (d, J = 2. 4 Hz, 1 H, H-6), 11. 26 (br s, 1 H, OH);
MS (FAB) m/z 345 (MH+).
- Methyl 2-Hydroxy-5-[4-(2-hydroxyethyl)piperazin-1-ylsulfonyl]benzoate
Example 3 EP1362858A1
Methyl 3-[4-(2-Hydroxyethyl)piperazin-1-ylsulfonyl]-6-n-propoxybenzoate
- To a mixture of methyl 2-hydroxy-5-(4-(2-hydroxyethyl)piperazin-1-ylsulfonyl)benzoate (800 mg, 2. 32 mmol) and K2CO3 (482 mg, 3. 49 mmol) in DMF (5 mL) was added 1-bromopropane (253 µL, 2.79 mmol), and the mixture was stirred at 60°C overnight. The reaction mixture was evaporated to dryness under reduced pressure, washed with H2O (10 mL), and the aqueous layer was further extracted with CH2Cl2 (50 mL x 2). The combined organic layer was dried (MgSO4), filtered, and the filtrate was evaporated to dryness under reduced pressure. The crude residue was purified by MPLC on silica gel (3% MeOH in CHCl3) to afford the titled compound (309 mg, 80%) as a white solid.
mp 88-89 °C (EtOAc/hexanes);
IR (neat) 3242 (OH), 1741 (C=O) cm-1;
1H NMR (CDCl3/TMS) δ 1. 09 (t, J = 7. 5 Hz, 3 H, OCH2CH2CH 3), 1. 84-1. 95 (m, 2 H, OCH2CH 2CH3), 2. 23 (br s, 1 H, CH2OH), 2. 54 (t, J= 5. 4 Hz, 2 H, NCH 2CH2O), 2. 60 (m, 4 H, 2 NCH2), 3. 04 (m, 4 H, 2 SO2NCH2), 3. 58 (t,J = 5. 4 Hz, 2 H, NCH2CH 2O), 3. 91 (s, 3 H, OCH3), 4. 08 (t, J= 6. 6 Hz, 2 H, OCH 2CH2CH3), 7. 07 (d, J = 9. 0 Hz, 1 H, H-3), 7. 82 (dd, J = 9. 0 Hz, 2. 4 Hz, 1 H, H-4), 8. 15 (d, J = 2. 4 Hz, 1 H, H-6);
MS (FAB) m/z 387 (MH+).
- FURTHER INFO OTHER THAN ABOVE PATENT
- HYDROLYSE Methyl 3-[4-(2-Hydroxyethyl)piperazin-1-ylsulfonyl]-6-n-propoxybenzoate TO -COOLi SALT using LiOH
- CONDENSE WITH 3-amino-1-ethyl-4-propyl-1H-pyrrole-2-carboxamide USING HOBt AND DMAP/ PYRIDINE
9……….Methyl 3-[4-(2-Hydroxyethyl)piperazin-1-ylsulfonyl]-6-n-propoxybenzoate R8= ME, R4=PROPYL, W=N, n=2
10……….3-amino-1-ethyl-4-propyl-1H-pyrrole-2-carboxamide R1=ETHYL, R2=H, R3=PROPYL, IN ABOVE
YOU WILL GET A COMPD
R1 ETHYL
R2=H
R3= PROPYL
R4 = PROPYL
W=N
n=2
IS MIRODENAFIL precursor ie n-1 compund
- CYCLIZE THIS WITH BuOK/tBuOH AND USE ACID TO GET FINAL PRODUCT MIRODENAFIL
- A cyclization reaction is generally carried out by heating at an elevated temperature, for example 50-150° C., in the presence of an acid or a base in a suitable solvent such as an aqueous C1-C4 alkanol, water, a halogenated hydrocarbon, or acetonitrile. Thus, for example, the cyclization may be affected by treatment of a compound with an inorganic or organic base such as sodium hydroxide, potassium carbonate or potassium tert-butoxide, in an alcoholic aqueous medium, preferably potassium tert-butoxide in tert-butanol at 60° C. to reflux temperature.
SYNTHESIS OF 1-(2-hydroxyethyl)piperazine needed for MIRODENAFIL SYNTHESIS
Compounds of the formula (29) can be prepared from the compounds of the formula (30):
wherein X and P are as previously defined.
note X=N ATOM, n = 2
…………………………….
MIRODENAFIL
Two methods were published for the determination of mirodenafil in biological fluids. Choi et al. (2009) describe an isocratic reversed-phase liquid chromatographic method for simultaneous analysis of mirodenafil and its two main metabolites, SK3541 and SK3544, in rat plasma, urine, and tissue homogenates. The authors used a simple deproteinization procedure for sample preparation, and the compounds were separated on a C18 column (250 mm x 4.6 mm, i.d.; 5 µm particle size; Shiseido, Tokyo, Japan). The mobile phase was constituted with 0.02 M ammonium acetate buffer (pH 6):acetonitrile (52:48, v/v) at a flow rate of 1.4 mL/min. UV detection was at 254 nm.
Lee et al. (2009) developed a study with the proposed method to determine sildenafil and mirodenafil in the plasma and corpus cavernosum tissue of rats using LC–MS/MS. A CapcellPak phenyl column (2.1mm x 150 mm, 5µm) maintained constant at 40 ºC was used for the separation. The mobile phase consisted of 90% acetonitrile in 5 mM ammonium formate (pH 6.0). A gradient program was used for the LC separation with a flow rate of 0.2 mL/min.
References
- Paick JS, Ahn TY, Choi HK, Chung WS, Kim JJ, Kim SC, Kim SW, Lee SW, Min KS, Moon KH, Park JK, Park K, Park NC, Suh JK, Yang DY, Jung HG (November 2008). “Efficacy and safety of mirodenafil, a new oral phosphodiesterase type 5 inhibitor, for treatment of erectile dysfunction”. The Journal of Sexual Medicine 5 (11): 2672–80. doi:10.1111/j.1743-6109.2008.00945.x. PMID 18638004.
- Kim BH, Yi S, Kim J, Lim KS, Kim KP, Lee B, Shin SG, Jang IJ, Yu KS (June 2009). “Influence of alcohol on the hemodynamic effects and pharmacokinetic properties of mirodenafil: a single-dose, randomized-sequence, open-label, crossover study in healthy male volunteers in Korea”.Clinical Therapeutics 31 (6): 1234–43. doi:10.1016/j.clinthera.2009.06.008. PMID 19695390.
- Shin KH, Kim BH, Kim TE, Kim JW, Yi S, Yoon SH, Cho JY, Shin SG, Jang IJ, Yu KS (December 2009). “The effects of ketoconazole and rifampicin on the pharmacokinetics of mirodenafil in healthy Korean male volunteers: an open-label, one-sequence, three-period, three-treatment crossover study”.Clinical Therapeutics 31 (12): 3009–20. doi:10.1016/j.clinthera.2009.12.012. PMID 20110038.
- Synthesis of 5-ethyl-2-[5-[4-(2-hydroxyethyl)piperazin-1-ylsulfonyl]-2-n-propoxyphenyl]-7-n-propyl-3,5-dihydro-4H-pyrrolo[3,2-d]-[2-14C]pyrimidin-4-one·2 HCl (14C-SK3530·2 HCl)J Label Compd Radiopharm 2006, 49(13): 1141
- More information about mirodenafil can be found at Paick J S et al., (2008) The Journal of Sexual Medicine, 5 (11): 2672-80.
- PDE-5 inhibitor that came into the market recently (Choi et al., 2009; Lee et al., 2009).not currently approved for use in the United States but clinical trials are being conducted.
- Crystal forms of SK-3530.
Song HO, Sohn YT.Arch Pharm Res. 2010 Dec;33(12):2033-6. doi: 10.1007/s12272-010-1220-3. Epub 2010 Dec 30. - Looking to the future for erectile dysfunction therapies.Hatzimouratidis K, Hatzichristou DG.Drugs. 2008;68(2):231-50. Review.
- Paick JS, Ahn TY, Choi HK, Chung WS, Kim JJ, Kim SC, Kim SW, Lee SW, Min KS, Moon KH, Park JK, Park K, Park NC, Suh JK, Yang DY, Jung HG (November 2008). “Efficacy and safety of mirodenafil, a new oral phosphodiesterase type 5 inhibitor, for treatment of erectile dysfunction”. The Journal of Sexual Medicine 5 (11): 2672–80. doi:10.1111/j.1743-6109.2008.00945.x. PMID 18638004.
- Kim BH, Yi S, Kim J, Lim KS, Kim KP, Lee B, Shin SG, Jang IJ, Yu KS (June 2009). “Influence of alcohol on the hemodynamic effects and pharmacokinetic properties of mirodenafil: a single-dose, randomized-sequence, open-label, crossover study in healthy male volunteers in Korea”. Clinical Therapeutics 31 (6): 1234–43.doi:10.1016/j.clinthera.2009.06.008. PMID 19695390.
- Shin KH, Kim BH, Kim TE, Kim JW, Yi S, Yoon SH, Cho JY, Shin SG, Jang IJ, Yu KS (December 2009). “The effects of ketoconazole and rifampicin on the pharmacokinetics of mirodenafil in healthy Korean male volunteers: an open-label, one-sequence, three-period, three-treatment crossover study”. Clinical Therapeutics 31 (12): 3009–20.doi:10.1016/j.clinthera.2009.12.012. PMID 20110038.
- Matheny, C., et al., Drug Metab. Dispos., 32, 1008 (2004)
Gupta, M., et al., J. Clin. Pharmacol., 45, 987 (2005)
Ek, M., et al., Biochem. Pharmacol., 74, 496 (2007)
Lee, H., et al., Xenobiotica, 38, 21 (2008)
PATENTS
1 WO 2001060825
2.WO 2013085276
3 KR 2013086771
4 WO2008/4796 A1
| WO2006018088A1 * | Jul 15, 2005 | Feb 23, 2006 | Switch Biotech Ag | Use of a pde 5 inhibitor for treating and preventing hypopigmentary disorders |
| KR20010083637A * | Title not available |
| US6962911 * | Feb 15, 2001 | Nov 8, 2005 | Sk Chemicals Co., Ltd. | Pyrrolopyrimidinone derivatives, process of preparation and use |
| US20100069632 * | Jul 3, 2007 | Mar 18, 2010 | Sk Chemicals Co., Ltd | Salts of pyrrolopyrimidinone derivatives and process for preparing the same |
| EP2038282A1 * | Jul 3, 2007 | Mar 25, 2009 | SK Chemicals, Co., Ltd. | Salts of pyrrolopyrimidinone derivatives and process for preparing the same |
.....................................................................................
6 LODENAFIL CARBONATE
Lodenafil carbonate
UNII-29X84F932D, CRIS-031
bis-(2-{4-[4-ethoxy-3-(1-methyl-7-oxo-3-propyl-6,7-dihydro-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-benzenesulfonyl]piperazin-1-yl}-ethyl)carbonate
5-{2-ethoxy-5-[(4-hydroxyethyl-1-piperazinyl)sulfonyl]phenyl}-1-methyl-3-n-propyl-1,6-dihydro-7H-pyrazole[4,3-d]pyrimidin-7-one. IS THE NAME OF MONOMER
398507-55-6 CAS
Cristalia (Originator)
| C47 H62 N12 O11 S2= MF | |
| Molecular Weight | 1035.199 |
Lodenafil is a drug belonging to a class of drugs called PDE5 inhibitor, which many other erectile dysfunction drugs such as sildenafil, tadalafil, and vardenafil also belong to. Like udenafil and avanafil it belongs to a new generation of PDE5 inhibitors.
- Sildenafil, tadalafil, vardenafil, and the newer udenafil and avanafil selectively inhibit PDE5, which is cGMP-specific and responsible for the degradation of cGMP in the corpus cavernosum. These phosphodiesterase inhibitors are used primarily as remedies for erectile dysfunction, as well as having some other medical applications such as treatment of pulmonary hypertension.
Lodenafil is formulated as a dimer, lodenafil carbonate, which breaks down in the body to form two molecules of the active drug lodenafil. This formulation has higher oral bioavailability than the parent drug.[1]
It is manufactured by Cristália Produtos Químicos e Farmacêuticos in Brazil and sold there under the brand-name Helleva.[2]
Helleva (Lodenafil Carbonate) is an oral PDE5 inhibitor prescribed to treat men suffering from erectile dysfunction. It operates by relaxing muscles and dilating blood vessels in the penis to increase circulation making it easier to attain and maintain an erection.
It has undergone Phase III clinical trials,[3][4][5] but is not yet approved for use in the United States by the U.S. Food and Drug Administration.
lodenafil
...........
SYNTHESIS
WO 2002012241 OR US7148350
MONOMER synthesis
PIPERAZINE
AND
ETHYL CHLORO ACETATE
WILL GIVE
Ethyl 1-piperazinylacetate
SEE RXN 1 BELOW
Reaction 1:
Synthesis of Piperazine Ethyl Acetate
To a reaction blend containing 100 g (3 Eq, 0.515 mol, MW=194) of piperazine, 26.3 mL (1.1 Eq, 0.189 mol, MW=101, d=0.726) of triethylamine in 200 mL of isopropanol, add to a solution previously prepared of 18.4 mL (1 Eq., 0.172 mol, MW=122.55, d=1.15) of chloroacetate of ethyl in 140 mL of isopropanol under stirring, at room temperature. Keep the reaction medium under stirring, monitoring the reaction termination by means of a chromatography of the thin layer (about 2–3 hours). Add a solution of 40.6 g (0.344 mol) of succinic acid in 140 mL of isopropanol. Keep the system under stirring for about 30 minutes to assure total precipitation of the succinate salt of piperazine formed. Filter this salt and concentrate the filtrate containing the mono and dialkyled derivatives. We obtain a slightly yellowish oil, which is used in later phases without purification.
Mass obtained=33 g
GC/MS: Monoalkylated derivative 72%, and dialkylated 22%.
NEXT
Piperazine Ethyl Acetate
AND
5-(5-chlorosulfonyl-2-ethoxyphenyl)-1-methyl-3n-propyl-1,6-dihydro-7H-pyrazole[4,3-d]pyrimidin-7-one
WILL REACT TO GIVE... 5-{2-ethoxy-5-[(4-ethyl acetate 1-piperazinyl)sulfonyl]phenyl}-1-methyl-3-n-propyl-1,6-di-hydro-7H-pyrazole[4,3-d]pyrimidin-7-one AS IN RXN 4 BELOW
Reaction 4:
Synthesis of 5-{2-ethoxy-5-[(4-ethyl acetate 1-piperazinyl)sulfonyl]phenyl}-1-methyl-3-n-propyl-1,6-di-hydro-7H-pyrazole[4,3-d]pyrimidin-7-one.
Suspend 24.6 g (60 mmol, MW=410.9) of 5-(5-chlorosulfonyl-2-etoxyphenyl)-1-methyl-3n-propyl-1,6-dihydro-7H-pyrazole[4,3-d]pyrimidin-7-one in 900 mL of ethanol absolute. Under stirring and at room temperature, add at only one time, a solution containing 31.0 g (3 Eq., 180 mmol MW=172) of N-piperazine ethyl acetate (Reaction 1) dissolved in 150 mL of ethanol absolute. In an interval of 2–10 minutes, all solid is consumed, forming a clean and homogeneous solution, and after that starts the precipitation of the expected product. At the end of the reaction, which lasts 2–3 hours (monitored by chromatography of thin layer), the product is vacuum filtered and the solid is washed with two portions of 50 mL of iced absolute ethanol. 29 g are obtained (yielding=89%) from the product as a white solid of MP=165.5–166.5° C.
Reaction 7:
Intermediate 1
5-{2-ethoxy-5-[(4-hydroxyethyl-1-piperazinyl)sulfonyl]phenyl}-1-methyl-3-n-propyl-1,6-dihydro-7H-pyrazole[4,3-d]pyrimidin-7-one. IS MONOMER
please note during LAH redn ............. the PIP CH2-C=O-O CH2 CH3 BECOMES PIP-CH2CH2-OH
To a suspension of lithium aluminum hydride (0.74 g 2.2 Eq. MW=37.9) in 25 mL of THF, slowly add, under stirring and at room temperature, a suspension of 5.0 g (9.1 mmol, MW=546.6) of 5-{2-ethoxy-5-[(4-ethyl acetate 1-piperazinyl)sulfonyl]phenyl}-1-methyl-3-n-propyl-1,6-di-hydro-7H-pyrazole[4,3-d]pyrimidin-7-one in 50 mL of THF. The system is maintained under stirring, monitoring the consumption of the product by chromatography of thin layer, until the complete consumption of the starting reagent (about 5–6 hours). Slowly add water to the reaction medium and, when there is no longer release of H2, add HCl 1M regulating pH for 7. Extract the product with 3 200 mL-portions of chloroform, dry with anhydrous sodium sulfate and vacuum concentrate the product. It is obtained 3.8 g of the product as a cream solid MP=183–187° C. yielding 83%. The same was crystallized from methanol and DMF yielding a slightly yellowish solid with melting point at 189–192° C.
note ............. the PIP CH2-C=O-O CH2 CH3 BECOMES PIP-CH2CH2-OH
HOMODIMER CARBONATE
EXAMPLE 1B
Homodimer Carbonate of Intermediate 1—Alternative Method
A phosgene solution (3.5 g, 35 mmol) dissolved in 20 mL of toluene was added dropwise to a solution of 2.02 g (4 mmol) of 5-{2-ethoxy-5-[(4-hydroxyethyl-1-piperazinyl)sulfonyl]phenyl}-1-methyl-3-n-propyl-1,6-dihydro-7H-pyrazole[4,3-d]pyrimidin-7-one, suspended in 44 mL of toluene. The reaction mixture resulting is stirred and followed by chromatography analysis of thin layer every hour until the reagent conversion in its chloroformate was completed. When the analysis indicates the complete consumption of 5-{2-ethoxy-5-[(4-hydroxyethyl-1-piperazinyl)sulfonyl]phenyl}-1-methyl-3-n-propyl-1,6-dihydro-7H-pyrazole[4,3-d]pyrimidin-7-one, the volatile compounds of the reaction are vacuum removed (solvents and phosgene), yielding the esther chloroformate raw derivative of 5-{2-ethoxy-5-[(4-hydroxyethyl-1-piperazinyl)sulfonyl]phenyl}-1-methyl-3-n-propyl-1,6-dihydro-7H-pyrazole[4,3-d]pyrimidin-7-one.
The raw chloroformate obtained above (4.0 mmol, 2.27 g) is dissolved in about 30 mL of dichloromethane, to which is added 2.07 g (4.1 mmol) of 5-{2-ethoxy-5-[(4-hydroxyethyl-1-piperazinyl)sulfonyl]phenyl}-1-methyl-3-n-propyl-1,6-dihydro-7H-pyrazole[4,3-d]pyrimidin-7-one, followed by the addition of 4 mL of dichloromethane containing 450 mg of triethylamine. The reaction mixture is maintained under stirring, being followed by chromatography of thin layer every hour until this indicates the end of the reaction (disappearing of chloroformate derivative). The reaction mixture is then diluted with 60 mL of dichloromethane, washed with NaCl saturated solution, after with sodium bicarbonate saturated solution and again with NaCl saturated solution. Organic phase is separated and dry with anhydrous sodium sulfate. The solvent is then evaporated to dry, yielding the dimer carbonate as a slightly yellowish solid.
This compound is re-crystallized from ethanol:DMF, yielding a pale white solid. Yielding m=3.2 g (76%)
Microanalysis: Theoretical C, (54.53%); H, (6.04%); N, (16.24%);
Obtained C, (54.45%); H, (6.02%); N, (16.17%).
INFO ABOUT INTERMEDIATE
5-(5-chlorosulfonyl-2-ethoxyphenyl)-1-methyl-3n-propyl-1,6-dihydro-7H-pyrazole[4,3-d]pyrimidin-7-one
| CAS No. | 139756-22-2 |
| Chemical Name: | 5-(5-Chlorosulfonyl-2-ethoxyphenyl)-1-methyl-3-propyl-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one |
| Synonyms: | Sildenafil Chlorosulfone IMpurity;Sildenafil Chlorosulfonyl IMpurity;5-(5-CHLOROSULFONYL-2-ETHOXY PHENYL)-1-METHYL-3-N-PROPYL-1;3-(6,7-dihydro-1-methyl-7-oxo-3-propyl-1 H-pyrazolo-(4-3-d)-pyrimidine-5;5-(5-Chlorosulfonyl-2-ethoxyphenyl)-1-methyl-3-propyl-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one;3-(4,7-Dihydro-1-methyl-7-oxo-3-propyl-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-4-ethoxy-benzenesulfonyl Chloride;4-Ethoxy-3-(1-Methyl-7-oxo-3-propyl-6,7-dihydro-1H-pyrazolo[4,3-d]pyriMidin-5-yl)benzene-1-sulfonyl chloride |
| CBNumber: | CB11175931 |
| Molecular Formula: | C17H19ClN4O4S |
..............
SYNTHESIS OF

2-butyrylamino-propionic acid
EXAMPLE 1A 2-Butyrylaminopropionic acid


22.27 g (250 mmol) of D,L-alanine and 55.66 g (550 mmol) of triethylamine are dissolved in 250 ml of dichloromethane, and the solution is cooled to 0° C. 59.75 g (550 mmol) of trimethylsilyl chloride are added dropwise, and the solution is stirred for 1 hour at room temperature and for 1 hour at 40° C. After cooling to −10° C., 26.64 g (250 mmol) of butyryl chloride are added dropwise, and the resulting mixture is stirred for 2 hours at −10° C. and for one hour at room temperature.
With ice-cooling, 125 ml of water are added dropwise and the reaction mixture is stirred at room temperature for 15 minutes. The aqueous phase is evaporated to dryness, the residue is titrated with acetone and the mother liquor is filtered off with suction. The solvent is removed and the residue is chromatographed. The resulting product is dissolved in 3N aqueous sodium hydroxide solution and the resulting solution is evaporated to dryness. The residue is taken up in conc. HCl and once more evaporated to dryness. The residue is stirred with acetone, precipitated solid is filtered off with suction and the solvent is removed under reduced pressure. This gives 28.2 g (71%) of a viscous oil which crystallizes after some time.
200 MHz 1H-NMR (DMSO-d6): 0.84, t, 3H; 1.22, d, 3H; 1.50, hex, 2H; 2.07, t, 2H; 4.20, quin., 1H; 8.09, d, 1H.
EXAMPLE 3A 2-Ethoxybenzonitrile


25 g (210 mmol) of 2-hydroxybenzonitrile are refluxed with 87 g of potassium carbonate and 34.3 g (314.8 mmol) of ethyl bromide in 500 ml of acetone overnight. The solid is filtered off, the solvent is removed under reduced pressure and the residue is distilled under reduced pressure. This gives 30.0 g (97%) of a colourless liquid.
200 MHz 1H-NMR (DMSO-d6): 1.48, t, 3H; 4.15, quart., 2H; 6.99, dt, 2H; 7.51, dt, 2H.
2-ethoxybenzamidine hydrochloride
EXAMPLE 4A 2-Ethoxybenzamidine hydrochloride


21.4 g (400 mmol) of ammonium chloride are suspended in 375 ml of toluene, and the suspension is cooled to 0° C. 200 ml of a 2M solution of trimethylaluminium in hexane are added dropwise, and the mixture is stirred at room temperature until the evolution of gas has ceased. After addition of 29.44 g (200 mmol) of 2-ethoxybenzonitrile, the reaction mixture is stirred at 80° C. (bath) overnight.
With ice-cooling, the cooled reaction mixture is added to a suspension of 100 g of silica gel and 950 ml of chloroform, and the mixture is stirred at room temperature for 30 minutes. The mixture is filtered off with suction, and the filter residue is washed with the same amount of methanol. The mother liquor is concentrated, the resulting residue is stirred with a mixture of dichloromethane and methanol (9:1), the solid is filtered off with suction and the mother liquor is concentrated. This gives 30.4 g (76%) of a colourless solid.
200 MHz 1H-NMR (DMSO-d6): 1.36, t, 3H; 4.12, quart., 2H; 7.10, t, 1H; 7.21, d, 1H; 7.52, m, 2H; 9.30, s, broad, 4H.
EXAMPLE 10A 2-(2-Ethoxy-phenyl)-5-methyl-7-propyl-3H-imidazo[5,1-f][1,2,4]triazin-4-one

7.16 g (45 mmol) of 2-butyrylamino-propionic acid and 10.67 g of pyridine are dissolved in 45 ml of THF and, after addition of a spatula tip of DMAP, heated to reflux. 12.29 g (90 mmol) of ethyl oxalyl chloride are slowly added dropwise, and the reaction mixture is refluxed for 3 hours. The mixture is poured into ice-water and extracted three times with ethyl acetate and the organic phase is dried over sodium sulphate and concentrated using a rotary evaporator. The residue is taken up in 15 ml of ethanol and refluxed with 2.15 g of sodium bicarbonate for 2.5 hours. The cooled solution is filtered.
With ice-cooling, 2.25 g (45 mmol) of hydrazine hydrate are added dropwise to a solution of 9.03 g (45 mmol) of 2-ethoxybenzamidine hydrochloride in 45 ml of ethanol, and the resulting suspension is stirred at room temperature for another 10 minutes. The ethanolic solution described above is added to this reaction mixture, and the mixture is stirred at a bath temperature of 70° C. for 4 hours. After filtration, the mixture is concentrated, the residue is partitioned between dichloromethane and water, the organic phase is dried over sodium sulphate and the solvent is removed under reduced pressure.
This residue is dissolved in 60 ml of 1,2-dichloroethane and, after addition of 7.5 ml of phosphorus oxychloride, refluxed for 2 hours. The mixture is diluted with dichloromethane and neutralized by addition of sodium bicarbonate solution and solid sodium bicarbonate. The organic phase is dried and the solvent is removed under reduced pressure. Chromatography using ethyl acetate and crystallization afford 4.00 g (28%) of a colourless solid, Rf=0.42 (dichloromethane/methanol=95:5)
200 MHz 1H-NMR (CDCl3): 1.02, t, 3H; 1.56, t, 3H; 1.89, hex, 2H; 2.67, s, 3H; 3.00, t, 2H; 4.26, quart., 2H; 7.05, m, 2H; 7.50, dt, 1H; 8.17, dd, 1H; 10.00, s, 1H.
EXAMPLE 15A 4-Ethoxy-3-(5-methyl-4-oxo-7-propyl-3,4-dihydro-imidazo[5,1-f][1,2,4]triazin-2-yl)-benzenesulphonyl chloride

At 0° C., 2.00 g (6.4 mmol) of 2-(2-ethoxy-phenyl)-5-methyl-7-propyl-3H-imidazo[5,1-f][1,2,4]triazin-4-one are slowly added to 3.83 ml of chlorosulphonic acid. At room temperature, the reaction mixture is stirred ovemight, and then poured into ice-water and extracted with dichloromethane. This gives 2.40 g (91%) of a colourless foam.
200 MHz 1H-NMR (CDCl3): 1.03, t, 3H; 1.61, t, 2H; 1.92, hex, 2H; 2.67, s, 3H; 3.10, t, 2H; 4.42, quart., 2H; 7.27, t, 1H; 8.20, dd, 1H; 8.67, d, 1H; 10.18, s, 1H.
Example 22 2-[2-Ethoxy-5-(4-hydroxyethyl-1-amino-piperazine-1-sulphonyl)-phenyl]-5-methyl-7-propyl-3H-imidazo[5,1-f][1,2,4]triazin-4-one
By the same method, starting with 0.04 g (0.097 mmol) of 4-ethoxy-3-(5-methyl-4-oxo-7-propyl-3,4-dihydro-imidazo[5,1-f][1,2,4]triazin-2-yl)-benzenesulphonyl chloride and 0.04 g (0.29 mmol) of 1-amino-4-hydroxyethylpiperazine, 46 mg (91%) of 2-[2-ethoxy-5-(4-hydroxyethyl-1-amino-piperazine-1-sulphonyl)-phenyl]-5-methyl-7-propyl-3H-imidazo[5,1-f][1,2,4]triazin-4-one are obtained.
Rf=0.08 (dichloromethane/methanol=19:1)
200 MHz 1H-NMR (CDCl3): 1.02, t, 3H; 1.59, t, 3H; 1.90, sex., 2H; 2.49, m, 6H; 2.62, s, 3H; 2.71, m, 4H; 3.00, t, 2H; 3.55, t, 2H; 4.31, quart., 2H; 7.14, d, 1H; 8.05, dd, 1H; 8.60, d, 1H.
..............
Methods of analysis
The development of lodenafil carbonate was reported by Toque et al. (2008). They observed the effects of lodenafil carbonate on rabbit and human corpus cavernosum relaxation, activity of PDE5 in human platelets, stability and metabolic studies in comparison with sildenafil and lodenafil, as well as the pharmacological evaluation of lodenafil carbonate after intravenous and oral administration in male beagles.
The determination of PDE activity, stability of lodenafil carbonate in human, dog and rat plasma and the pharmacokinetic parameters after a single intravenous or oral dose was carried out by LC-MS/MS analysis
Codevilla et al. (2011a) developed a stability-indicating reversed-phase liquid chromatography method using ultraviolet (UV) detection for the quantitative determination of lodenafil carbonate in tablets. The method can be useful for routine quality control assay and stability studies.
Another study for the determination of lodenafil carbonate in tablets was developed by Codevilla et al. (2011b). As an alternative to the LC method the authors suggested a UV-spectrophotometric method for the analysis of lodenafil carbonate in pharmaceutical form. The UV method offers advantages over other analytical methods due to its rapidity, simplicity, and lower cost. Recently, Codevilla et al. (2012) developed and validated a capillary zone electrophoresis (CZE) method for determination of lodenafil carbonate in drug products. There are some advantages to use the CZE method, such as rapid analysis, small sample and reagent consumption, high separation efficiency (Furlanetto et al., 2001; Yang et al., 2010). The results obtained from the UV-spectrophotometric method and CZE method were compared statistically with the LC method (Codevilla et al., 2011a) and the results showed no significant difference between these methods.
References
- Toque HA, Teixeira CE, Lorenzetti R, Okuyama CE, Antunes E, De Nucci G (September 2008). "Pharmacological characterization of a novel phosphodiesterase type 5 (PDE5) inhibitor lodenafil carbonate on human and rabbit corpus cavernosum". European Journal of Pharmacology 591 (1–3): 189–95. doi:10.1016/j.ejphar.2008.06.055. PMID 18593576.
- Cristália Product page. Retrieved on September 16, 2009.
- ukmedix Lodenafil article. Retrieved on September 16, 2009.
- Glina S, Toscano I, Gomatzky C, de Góes PM, Júnior AN, Claro JF, Pagani E (February 2009). "Efficacy and tolerability of lodenafil carbonate for oral therapy in erectile dysfunction: a phase II clinical trial". The Journal of Sexual Medicine 6 (2): 553–7. doi:10.1111/j.1743-6109.2008.01079.x.PMID 19040623.
- Glina S, Fonseca GN, Bertero EB, Damião R, Rocha LC, Jardim CR, Cairoli CE, Teloken C, Torres LO, Faria GE, da Silva MB, Pagani E (February 2010). "Efficacy and Tolerability of Lodenafil Carbonate for Oral Therapy of Erectile Dysfunction: A Phase III Clinical Trial". The Journal of Sexual Medicine 7 (5): 1928–1936. doi:10.1111/j.1743-6109.2010.01711.x. PMID 20214718.
- Toque H A et al., (2008) European Journal of Pharmacology, 591(1-3):189-95.
- Exploring the role of PDE5 inhibition in the treatment of muscular dystrophy
Drugs Fut 2011, 36(4): 321
..............................................................................
7 VARDENAFIL
VARDENAFIL
224785-90-4 CAS NO
Vardenafil hydrochloride (CAS NO.224785-91-5)
| Formula | C23H32N6O4S |
|---|---|
| Mol. mass | 488.604 g/mol |
4-[2-Ethoxy-5-(4-ethylpiperazin-1-yl)sulfonyl-phenyl]-9-methyl-7-propyl-3,5,6,8-tetrazabicyclo[4.3.0]nona-3,7,9-trien-2-one
Vivanza, Vardenafil (INN), Levitra (TN), STK642629, , LEVITRA
Vardenafil (INN) is a PDE5 inhibitor used for treating erectile dysfunction that is sold under the trade names Levitra (Bayer AG, GSK, and SP) andStaxyn.
Vardenafil was co-marketed by Bayer Pharmaceuticals, GlaxoSmithKline, and Schering-Plough under the trade name Levitra. As of 2005, the co-promotion rights of GSK on Levitra have been returned to Bayer in many markets outside the U.S. In Italy, Bayer sells vardenafil as Levitra and GSK sells it as Vivanza. Thus, because of European Union trade rules, parallel imports might result in Vivanza sold next to Levitra in the EU.
Vardenafil (Levitra) is an oral therapy for the treatment of erectile dysfunction. It is a selective inhibitor of cyclic guanosine monophosphate (cGMP)-specific phosphodiesterase type 5 (PDE5). Penile erection is a hemodynamic process initiated by the relaxation of smooth muscle in the corpus cavernosum and its associated arterioles. During sexual stimulation, nitric oxide is released from nerve endings and endothelial cells in the corpus cavernosum. Nitric oxide activates the enzyme guanylate cyclase resulting in increased synthesis of cyclic guanosine monophosphate (cGMP) in the smooth muscle cells of the corpus cavernosum. The cGMP in turn triggers smooth muscle relaxation, allowing increased blood flow into the penis, resulting in erection. The tissue concentration of cGMP is regulated by both the rates of synthesis and degradation via phosphodiesterases (PDEs). The most abundant PDE in the human corpus cavernosum is the cGMPspecific phosphodiesterase type 5 (PDE5); therefore, the inhibition of PDE5 enhances erectile function by increasing the amount of cGMP.
An orally disintegrating form, marketed as Staxyn, has been gaining approvals in countries such as the United States[1] and Canada.[2]
Vardenafil’s indications and contra-indications are the same as with other PDE5 inhibitors; it is closely related in function to sildenafil citrate (Viagra) and tadalafil (Cialis). The difference between the vardenafil molecule and sildenafil citrate is a nitrogen atom’s position and the change of sildenafil’spiperazine ring methyl group to an ethyl group. Tadalafil is structurally different from both sildenafil and vardenafil. Vardenafil’s relatively short effective time is comparable to but somewhat longer than sildenafil’s.
Beyond its indications for erectile dysfunction, vardenafil may be effective in the treatment of premature ejaculation, where it may significantly increase the time from vaginal penetration to ejaculation.[3]
The common, adverse drug reactions (side-effects) are the same as with other PDE5 inhibitors. The frequent vardenafil-specific side-effect is nausea; the infrequent side-effects are abdominal pain, back pain, photosensitivity, abnormal vision, eye pain, facial edema, hypotension, palpitation,tachycardia, arthralgia, myalgia, rash, itch, and priapism.
One possibly serious, but rare, side-effect with vardenafil is heart attack. Also, in rare cases, vardenafil use may cause priapism, a very painful emergency condition that can cause impotence if left untreated.[4]
On 18 October 2007, the U.S. Food and Drug Administration (FDA) announced that a warning about possible deafness (sudden hearing loss) would be added to the drug labels of Vardenafil, and other PDE5 inhibitors.[5]
Vardenafil, as with all PDE5 inhibitors, should not be used by men taking nitrate medications, because combining them with vardenafil might provoke potentially life-threatening hypotension (low blood pressure).
Further, Vardenafil causing lengthening of the QT interval. Therefore it should not be taken by men taking other medications that affect the QT interval (such as amiodarone).

Levitra 20mg Oral Tablet
It is available in 2.5 mg, 5 mg, 10 mg, and 20 mg doses in round orange tablets. The normal starting dose is 10 mg (roughly equivalent to 50 mg of sildenafil). Vardenafil should be taken 1 to 2 hours prior to sexual activity, with a maximum dose frequency of once per day. In some territories, such as the UK, only certain doses may be available.
Vardenafil is also available under the name Staxyn as a tablet which dissolves on the tongue rather than being swallowed in the form of a pill.
STAXYN is an oral therapy for the treatment of erectile dysfunction. This monohydrochloride salt of vardenafil is a selective inhibitor of cyclic guanosine monophosphate (cGMP)-specific PDE5.
Vardenafil HCl is designated chemically as piperazine, 1-[[3-(1,4-dihydro-5-methyl-4-oxo-7-propylimidazo[5,1f][1,2,4]triazin-2-yl)-4-ethoxyphenyl]sulfonyl]-4-ethyl-, monohydrochloride and has the following structural formula:
 |
Vardenafil HCl is a nearly colorless, solid substance with a molecular weight of 579.1 g/mol and a solubility of 0.11 mg/mL in water.
LEVITRA
TRIHYDRATE, HCL SALT
vardenafil hydrochloride is piperazine, 1-[[3-(1,4-dihydro-5-methyl-4-oxo-7-propylimidazo[5,1-f][1,2,4]triazin-2-yl)-4-ethoxyphenyl]sulfonyl]-4-ethyl-, mono -hydrochloride and can be structurally represented by Formula I.

The monohydrochloride salt of vardenafil is a selective inhibitor of cyclic guaosine monophosphate (cGMP)-specific phosphodiesterase type 5 (PDE5). It is commercially available in products sold under the brand name LEVITRA formulated as 2.5 mg, 5 mg, 10 mg, 20 mg film-coated tablets.
U.S. Pat. No. 6,362,178 B1 discloses vardenafil, its related compounds and processes for their preparation. The patent describes a process in which vardenafil is obtained by recrystallization in ether in Example 19. Vardenafil produced as per Example 19 is hereinafter referred as “crystalline Form I” of vardenafil. The patent also describes processes for the preparation of its monohydrochloride and dihydrochloride salts, which are formed in a combination of ether and dichloromethane. The patent also describes a process for the preparation of vardenafil monohydrochloride trihydrate.
U.S. Patent Application Publication No. 2005/0203298 also describes a process for the preparation of vardenafil, and its monohydrochloride trihydrate.
Chemical synthesis of vardenafil has mostly been directed to the preparation of the trihydrate of monohydrochloride of vardenafil.
In WO 99/24433, sulphonamide-substituted imidazotriazinones are described as potent inhibitors of either one or more of the cyclic guanosine 3′,5′-monophosphate-metabolizing phosphodiesterases (cGMP PDEs). According to the nomenclature of Beavo and Reifsnyder (Trends in Pharmacol. Sci. 11, 150-155, 1990), these cGMP PDEs are the phosphodiesterase isoenzymes PDE-I, PDE-II and PDE-V.
According to WO 99/24433, the sulphonamide-substituted imidazotriazinones described therein are prepared from corresponding 2-ethoxyphenyl-substituted imidazotriazinones by reaction with chlorosulphonic acid and subsequent reaction with an appropriate amine, as is illustrated by the following scheme (R1 to R6 here have the meanings indicated in WO 99/24433):
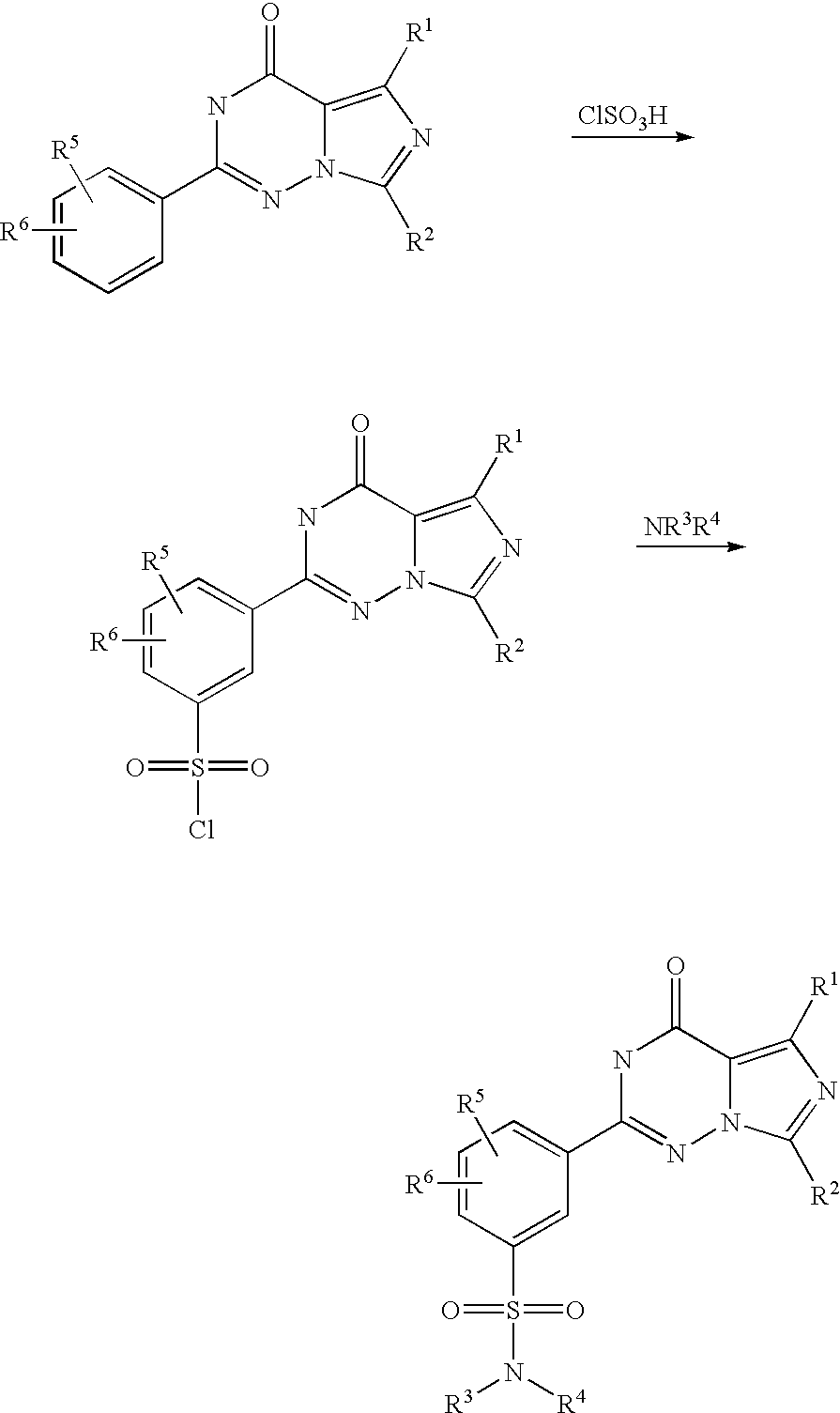
In this process, highly reactive chlorosulphonic acid has to be used as a reagent. Moreover, the imidazotriazinonesulphonyl chlorides formed as intermediates are sensitive to hydrolysis, which, in particular in the conversion of this preparation process to the industrial scale, can lead to not inconsiderable yield variations.
It was therefore the object of the present invention to make available a process for the preparation of sulphonamide-substituted imidazotriazinones in which the disadvantages of the above process known from the prior art are avoided.
This object is achieved according to the present invention by a process as in claim 1. In particular, in the process according to the invention as in claim 1 the use of chlorosulphonic acid is avoided by introduction of the sulphonic acid via a reaction with sulphuric acid and subsequent reaction with thionyl chloride. Moreover, the reaction with thionyl chloride and the subsequent reaction with an amine is carried out in a one-pot process, so that the imidazotriazinonesulphonyl chloride intermediate, which is sensitive to hydrolysis, does not need to be isolated. By means of this, yield variations on account of partial hydrolysis of this intermediate can be excluded. As a result of these advantages, the process according to the invention is much simpler to carry out on the industrial scale than the process described in WO 99/24433.
………………….
SYNTHESIS
2-butyrylamino-propionic acid
EXAMPLE 1A 2-Butyrylaminopropionic acid
22.27 g (250 mmol) of D,L-alanine and 55.66 g (550 mmol) of triethylamine are dissolved in 250 ml of dichloromethane, and the solution is cooled to 0° C. 59.75 g (550 mmol) of trimethylsilyl chloride are added dropwise, and the solution is stirred for 1 hour at room temperature and for 1 hour at 40° C. After cooling to −10° C., 26.64 g (250 mmol) of butyryl chloride are added dropwise, and the resulting mixture is stirred for 2 hours at −10° C. and for one hour at room temperature.
With ice-cooling, 125 ml of water are added dropwise and the reaction mixture is stirred at room temperature for 15 minutes. The aqueous phase is evaporated to dryness, the residue is titrated with acetone and the mother liquor is filtered off with suction. The solvent is removed and the residue is chromatographed. The resulting product is dissolved in 3N aqueous sodium hydroxide solution and the resulting solution is evaporated to dryness. The residue is taken up in conc. HCl and once more evaporated to dryness. The residue is stirred with acetone, precipitated solid is filtered off with suction and the solvent is removed under reduced pressure. This gives 28.2 g (71%) of a viscous oil which crystallizes after some time.
200 MHz 1H-NMR (DMSO-d6): 0.84, t, 3H; 1.22, d, 3H; 1.50, hex, 2H; 2.07, t, 2H; 4.20, quin., 1H; 8.09, d, 1H.
EXAMPLE 3A 2-Ethoxybenzonitrile
25 g (210 mmol) of 2-hydroxybenzonitrile are refluxed with 87 g of potassium carbonate and 34.3 g (314.8 mmol) of ethyl bromide in 500 ml of acetone overnight. The solid is filtered off, the solvent is removed under reduced pressure and the residue is distilled under reduced pressure. This gives 30.0 g (97%) of a colourless liquid.
200 MHz 1H-NMR (DMSO-d6): 1.48, t, 3H; 4.15, quart., 2H; 6.99, dt, 2H; 7.51, dt, 2H.
2-ethoxybenzamidine hydrochloride
EXAMPLE 4A 2-Ethoxybenzamidine hydrochloride
21.4 g (400 mmol) of ammonium chloride are suspended in 375 ml of toluene, and the suspension is cooled to 0° C. 200 ml of a 2M solution of trimethylaluminium in hexane are added dropwise, and the mixture is stirred at room temperature until the evolution of gas has ceased. After addition of 29.44 g (200 mmol) of 2-ethoxybenzonitrile, the reaction mixture is stirred at 80° C. (bath) overnight.
With ice-cooling, the cooled reaction mixture is added to a suspension of 100 g of silica gel and 950 ml of chloroform, and the mixture is stirred at room temperature for 30 minutes. The mixture is filtered off with suction, and the filter residue is washed with the same amount of methanol. The mother liquor is concentrated, the resulting residue is stirred with a mixture of dichloromethane and methanol (9:1), the solid is filtered off with suction and the mother liquor is concentrated. This gives 30.4 g (76%) of a colourless solid.
200 MHz 1H-NMR (DMSO-d6): 1.36, t, 3H; 4.12, quart., 2H; 7.10, t, 1H; 7.21, d, 1H; 7.52, m, 2H; 9.30, s, broad, 4H.
EXAMPLE 10A 2-(2-Ethoxy-phenyl)-5-methyl-7-propyl-3H-imidazo[5,1-f][1,2,4]triazin-4-one

7.16 g (45 mmol) of 2-butyrylamino-propionic acid and 10.67 g of pyridine are dissolved in 45 ml of THF and, after addition of a spatula tip of DMAP, heated to reflux. 12.29 g (90 mmol) of ethyl oxalyl chloride are slowly added dropwise, and the reaction mixture is refluxed for 3 hours. The mixture is poured into ice-water and extracted three times with ethyl acetate and the organic phase is dried over sodium sulphate and concentrated using a rotary evaporator. The residue is taken up in 15 ml of ethanol and refluxed with 2.15 g of sodium bicarbonate for 2.5 hours. The cooled solution is filtered.
With ice-cooling, 2.25 g (45 mmol) of hydrazine hydrate are added dropwise to a solution of 9.03 g (45 mmol) of 2-ethoxybenzamidine hydrochloride in 45 ml of ethanol, and the resulting suspension is stirred at room temperature for another 10 minutes. The ethanolic solution described above is added to this reaction mixture, and the mixture is stirred at a bath temperature of 70° C. for 4 hours. After filtration, the mixture is concentrated, the residue is partitioned between dichloromethane and water, the organic phase is dried over sodium sulphate and the solvent is removed under reduced pressure.
This residue is dissolved in 60 ml of 1,2-dichloroethane and, after addition of 7.5 ml of phosphorus oxychloride, refluxed for 2 hours. The mixture is diluted with dichloromethane and neutralized by addition of sodium bicarbonate solution and solid sodium bicarbonate. The organic phase is dried and the solvent is removed under reduced pressure. Chromatography using ethyl acetate and crystallization afford 4.00 g (28%) of a colourless solid, Rf=0.42 (dichloromethane/methanol=95:5)
200 MHz 1H-NMR (CDCl3): 1.02, t, 3H; 1.56, t, 3H; 1.89, hex, 2H; 2.67, s, 3H; 3.00, t, 2H; 4.26, quart., 2H; 7.05, m, 2H; 7.50, dt, 1H; 8.17, dd, 1H; 10.00, s, 1H.
EXAMPLE 15A 4-Ethoxy-3-(5-methyl-4-oxo-7-propyl-3,4-dihydro-imidazo[5,1-f][1,2,4]triazin-2-yl)-benzenesulphonyl chloride
At 0° C., 2.00 g (6.4 mmol) of 2-(2-ethoxy-phenyl)-5-methyl-7-propyl-3H-imidazo[5,1-f][1,2,4]triazin-4-one are slowly added to 3.83 ml of chlorosulphonic acid. At room temperature, the reaction mixture is stirred ovemight, and then poured into ice-water and extracted with dichloromethane. This gives 2.40 g (91%) of a colourless foam.
200 MHz 1H-NMR (CDCl3): 1.03, t, 3H; 1.61, t, 2H; 1.92, hex, 2H; 2.67, s, 3H; 3.10, t, 2H; 4.42, quart., 2H; 7.27, t, 1H; 8.20, dd, 1H; 8.67, d, 1H; 10.18, s, 1H.
Example 19 2-[2-Ethoxy-5-(4-ethyl-piperazine-1-sulphonyl)-phenyl]-5-methyl-7-propyl-3H-imidazo[5,1-f][1,2,4]triazin-4-one

470 mg (1.14 mmol) of 4-ethoxy-3-(5-methyl-4-oxo-7-propyl-3,4-dihydro-imidazo[5,1-f][1,2,4]triazin-2-yl)-benzenesulphonyl chloride are dissolved in 20 ml of dichloromethane and cooled to 0° C. 390 mg (3.42 mmol) of N-ethylpiperazine are added, and the reaction mixture is stirred at room temperature overnight. The mixture is diluted with dichloromethane, the organic phase is washed twice with water and dried over sodium sulphate and the solvent is removed under reduced pressure. Crystallization from ether gives 370 mg (66%) of a colourless solid.
400 MHz 1H-NMR (CDCl3): 1.01, t, 3H; 1.59, t, 3H; 1.88, hex, 2H; 2.42, quart., 2H; 2.56, m, 4H; 2.63, s, 3H; 3.00, t, 2H; 3.10, m, 4H; 4.33, quart., 2H, 7.17, d, 1H; 7.88, dd, 1H; 8.44, d, 1H; 9.75, s, 1H.
…………………….
EXAMPLE 7 Preparation of the Trihydrate of Vardenafil Monohydrochloride
14 g of vardenafil hydrochloride was taken into a round bottom flask followed by the addition of 70 ml water and the pH of the reaction mass was adjusted using sodium hydroxide to 11 at 30° C. 280 ml of dichloromethane was added to the above reaction mass and the layers were separated. The organic layer was dried over sodium sulfate and the organic layer was transferred into a round bottom flask and subjected to heating for distillation at 40° C. for 1.5 hours. The solid material was transferred into a round bottom flask and 36 ml of a mixture of acetone and water in 12:1 ratio was added with stirring, then 2.2 ml of 36% aqueous hydrochloric acid was added with stirring. The reaction mass was heated to a temperature of about 45° C. and the undissolved particles were removed by filtration. The filtrate was taken into a round bottom flask and cooled to 5° C., maintained for 45 minutes at 3 to 5° C. followed by the filtration of the solid which was then subjected to suction drying and finally dried at 40° C. to yield 9.0 g of the trihydrate of vardenafil monohydrochloride.
……………………..
STARTING COMPOUNDS
Example I Preparation of 2-(2-ethoxyphenyl)-5-methyl-7-propyl-3H-imidazo-[5,1-f][2,4]triazin-4-oneIa) Preparation of 2-butyrylaminopropionic acid

A solution of 100 kg of D,L-alanine in aqueous sodium hydroxide solution is reacted in the cold with 119 kg of butyryl chloride. After addition of butyl acetate, the mixture is acidified with hydrochloric acid, the organic phase is separated off and the aqueous phase is re-extracted. The organic phase is dried by azeotropic distillation. The crystallizate is isolated, washed with butyl acetate and dried.
Yield: 132.6 kg (68%)
1H-NMR: δ=0.8 (t, 3H), 1.25 (d, 3H), 1.5 (m, 2H), 2.1 (t, 2H), 4.2 (q, 1H), 8.1 (d, NH), 12.0-12.7 (s, COOH)
MS: 336 (2M+NH4, 40), 319 (2M+H, 15), 177 (M+NH4, 100), 160 (M+H, 20)
Ib) Preparation of 2-ethoxybenzonitrile

260 kg of thionyl chloride are added at 85-95° C. to a suspension of 250 kg of 2-ethoxybenzamide in toluene under metering control. The reaction mixture is stirred in the presence of heat. Thionyl chloride and toluene are then distilled off in vacuo. The product is employed in the subsequent stage as a crude product.
Yield: 228.5 kg (crude product)
1H-NMR: δ=1.45 (t, 3H), 4.15 (q, 2H), 7.0 (m, 2H, phenyl), 7.5 (m, 2H, phenyl)
MS: 312 (2M+N4, 35), 165 (M+NH4, 100), 147 (5)
Ic) Preparation of 2-ethoxy-N-hydroxybenzamidine

111 kg of 2-ethoxybenzonitrile (crude product) from Example Ib are heated under reflux with 164 1 of triethylamine and 73 kg of hydroxylamine hydrochloride in isopropanol. The reaction mixture is treated with water and cooled. The crystallizate is isolated, washed and employed in the subsequent stage as a moist product.
Yield: 92.6 kg (moist product)
1H-NMR: δ=1.35 (t, 3H), 4.1 (q, 2H), 5.6 (s, 2H), 6.9-7.4 (4H, phenyl), 9.4 (s, 1H, OH)
MS: 361 (2M+H, 30), 198 (M+N, 30), 181 (M+H, 100)
Id) Preparation of 2-ethoxybenzamidine hydrochloride

135 kg of 2-ethoxy-N-hydroxybenzamidine (moist product) from Example Ic are hydrogenated at 50-60° C. in acetic acid using palladium on carbon as a catalyst. For the work-up, the hydrogenation reaction is freed from the catalyst, treated with hydrochloric acid and concentrated. Residual acetic acid and water are removed by azeotropic distillation with toluene. The crystallizate is isolated and dried in vacuo.
Yield: 136.4 kg
H-NMR: 1.35 (t, 3H), 4.15 (q, 2H), 7.1-7.7 (m, 4H, phenyl), 9.1-9.4 (2×s, 3H), 10.5-10.7 (s, 1H)
MS: 329 (2M+H, 10), 165 (M+H, 100)
Ie) Preparation of 2-(2-ethoxyphenyl)-5-methyl-7-propyl-3H-imidazo[5,1-f][1,2,4]-triazin-4-one

231 kg of 2-butyrylaminopropionic acid from Example Ia are treated in tetrahydrofuran with 341 kg of pyridine, catalytic amounts of 4-N,N-dimethylaminopyridine and 392 kg of ethyl chloroxalate and stirred with heating under reflux. The reaction mixture is taken up in ethyl acetate, washed with water and the ethyl acetate phase is concentrated. The distillation residue is taken up in methanol and reacted with the following solution.
192 kg of 2-ethoxybenzamidine hydrochloride from Example Id are treated in methanol with 47.5 kg of hydrazine hydrate and the mixture is stirred at room temperature. The solution is combined with the solution of 2-butyrylamino-1-ethoxycarbonylpropenyl ethyl oxalate prepared above. The reaction mixture thus obtained is stirred with heating under reflux. Methanol is removed by distillation and replaced by acetic acid.
Option A:
138.6 kg of phosphorus oxychloride are added and stirred in the presence of heat.
Acetic acid is distilled off in vacuo. The residue is treated with water and dichloromethane or optionally methyl isobutyl ketone and rendered neutral using sodium hydroxide solution. The organic phase is concentrated, and the residue is dissolved in acetone and crystallized with cooling. The crystallizate is isolated, washed and dried.
Option B:
At least 190 kg of acetyl chloride are added and stirred in the presence of heat. Acetic acid is distilled off in vacuo. The distillation residue is treated with acetone and water, and the product is crystallized by rendering neutral with sodium hydroxide solution. The product is isolated, washed and dried.
Yield: 90-160 kg
1H-NMR: δ=1.0 (t, 3H), 1.6 (t, 3H), 1.9 (m, 2H), 2.8 (s, 3H), 3.3 (t, 2H), 4.3 (q, 2H), 7.0-8.2 (Ar, 4H), 10.3 (CONH, 1H)
MS: 313 (M+H, 100), 149 (25), 151 (40), 121 (15)
HPLC: Kromasil C-18 phase, neutral phosphate buffer, acetonitrile, 233 nm, linear gradient of 30% acetonitrile ->80% acetonitrile (30 min.): 99 area % (Rt 19.1)
PREPARATION EXAMPLES Example 1a 4-ethoxy-3-(5-methyl-4-oxo-7-propyl-3,4-dihydroimidazo[5,1-fl-][1,2,4]triazin-2-yl)benzenesulphonic acid

194 kg of 2-(2-ethoxyphenyl)-5-methyl-7-propyl-3H-imidazo[5,1-f][1,2,4]triazin-4-one from Example Ie are reacted with 504 kg of concentrated sulphuric acid. The reaction mixture is added to water, cooled, and the crystallizate is isolated and dried in vacuo.
Yield: 195.2 kg
1H-NMR: δ=0.95 (t, 3H), 1.3 (t, 3H), 1.8 (m, 2H), 2.6 (s, 3H), 3.05 (t, 2H), 4.1 (q, 2H), 7.15 (Ar, 1H), 7.75 (m, 2H), 12.3 (SO2OH)
MS: 393 (M+H, 100), 365 (25), 151 (40)
HPLC: X-Terra C-18 phase, aqueous phosphoric acid, acetonitrile, 242 nm, linear gradient of 10% acetonitrile ->90% acetonitrile (20 min.):
98 area % (R, 9.2)
Example 1b) 2-[2-ethoxy-5-(4-ethlylpiperazin-1-sulphonyl)phenyl]-5-methyl-7-propyl-3H-imidazo[5,1-f][1,2,4]triazin-4-one

22.5 kg of 4-ethoxy-3-(5-methyl-4-oxo-7-propyl-3,4-dihydro-imidazo[5,1-f][1,2,4]-triazin-2-yl)benzenesulphonic acid from Example 1a are reacted with 74 kg of thionyl chloride and catalytic amounts of dimethylformamide until the evolution of gas has ended. Xylene is repeatedly added to the reaction mixture and thionyl chloride is distilled off. 15.1 kg of N-ethylpiperazine are added to the suspension and it is stirred. After the addition of water, it is adjusted to pH 1 using hydrochloric acid, and the phases are separated. The aqueous phase is treated with acetone and rendered neutral by addition of sodium hydroxide solution. The mixture is cooled, and the crystallizate is isolated, washed and dried in vacuo.
Yield: 26.1 kg
1H-NMR: δ=1.0 (2×t, 6H), 1.6 (t, 3H), 1.9 (m, 2H), 2.45 (q, 2H), 2.55 (m, 4H), 2.65 (s, 3H), 3.0 (t, 2H), 3.1 (m, 4H), 4.35 (q, 2H), 7.15 (Ar, 1H), 7.9 (Ar, 1H), 8.4 (Ar, 1H), 9.8 (CONH)
MS: 489 (M+H, 100), 345 (10), 313, (10), 285 (10), 113 (20)
HPLC: X-Terra C-18 phase, neutral phosphate buffer, acetonitrile, 242 nm, linear gradient of 20% acetonitrile ->75% acetonitrile (20 min.): 98 area % (Rt 16.3)
1 c) 2-[2-ethoxy-5-(4-ethylpiperazin-1-sulphonyl)phenyl]-5-methyl-7-propyl-3H-imidazo[5,1-fl][1,2,4]triazin-4-one hydrochloride trihydrate

22.5 kg of 2-[2-ethoxy-5-(4-ethylpiperazin-1-sulphonyl)phenyl]-5-methyl-7-propyl-3H-imidazo[5,1-f][1,2,4]triazin-4-one from Example 1b are dissolved in 5.1 kg of concentrated hydrochloric acid and acetone/water (12:1 v/v) in the presence of heat. The clear solution is filtered hot and crystallized by cooling and seeding. The crystallizate is isolated, washed and dried in vacuo at about 30° C. and about 300 mbar.
Yield: 25.4 kg
M.p. (DSC): 192° C.
HPLC: X-Terra C-18 phase, neutral phosphate buffer, acetonitrile, 242 nm, linear gradient of 20% acetonitrile ->75% acetonitrile (20 min.): 99 area % (Rt 16.3)
- http://www.pharmpro.com/News/Feeds/2010/06/pharmaceutical-companies-bayer-new-erectile-dysfunction-treatment-staxyn-approve/
- http://www.newswire.ca/en/story/832217/staxyn-new-innovation-in-erectile-dysfunction-helps-younger-men-rise-to-the-occasion
- A Aversa et al. “Effects of vardenafil administration on intravaginal ejaculatory latency time in men with lifelong premature ejaculation”. Retrieved 2010-12-14.
- Schools of Pharmacy (Glen L. Stimmel, Pharm.D., and Mary A. Gutierrez, Pharm.D.) and Medicine (Glen L. Stimmel, Pharm.D.), University of Southern California, Los Angeles, California. “Counseling Patients About Sexual Issues: Drug-Induced Priapism”. Medscape. Retrieved 2010-12-06.
- “FDA Announces Revisions to Labels for Cialis, Levitra and Viagra”. Food and Drug Administration. 2007-10-18. Retrieved 2009-08-06.
- Official Levitra website
- PubChem Information
- 2-1-2013Synthesis of quinoline derivatives: discovery of a potent and selective phosphodiesterase 5 inhibitor for the treatment of Alzheimer’s disease.European journal of medicinal chemistry
PATENTS
| US6362178 * | Oct 31, 1998 | Mar 26, 2002 | Bayer Aktiengesellschaft | 2-phenyl substituted imidazotriazinones as phosphodiesterase inhibitors |
| US20050203298 * | May 5, 2005 | Sep 15, 2005 | Bayer Healthcare Aktiengesellschaft | Process for the preparation of sulphonamide-substituted imidazotriazinones |
| US20060111354 * | Jul 3, 2003 | May 25, 2006 | Peter Serno | Medicaments containing vardenafil hydrochloride trihydrate |
| WO2004006894A1 * | Jul 3, 2003 | Jan 22, 2004 | Bayer Healthcare Ag | Medicaments containing vardenafil hydrochloride trihydrate |
11-4-2011
|
ROFLUMILAST FOR THE TREATMENT OF DIABETES MELLITUS
| |
9-14-2011
|
Roflumilast for the Treatment of Diabetes Mellitus
| |
8-5-2011
|
N-BUTYRAMIDE, THE PREPARATION METHOD AND USE THEREOF
| |
3-4-2011
|
Fatty Acid Oxidation Inhibitors Treating Hyperglycemia and Related Disorders
| |
1-14-2011
|
2-PHENYL SUBSTITUTED IMIDAZOTRIAZINONES AS PHOSPHODIESTERASE INHIBITORS
| |
9-17-2010
|
SUBSTITUTED PDE5 INHIBITORS
| |
7-16-2010
|
Combination treatment for diabetes mellitus
| |
4-28-2010
|
2-Phenyl substituted imidazotriazinones as phosphodiesterase inhibitors
| |
4-14-2010
|
2-PHENYL SUBSTITUTED IMIDAZOTRIAZINONES AS PHOSPHODIESTERASE INHIBITORS
| |
2-5-2010
|
Heterocyclic Compounds And Uses Thereof In The Treatment Of Sexual Disorders
|
12-25-2009
|
Therapeutic Compositions Comprising a Specific Endothelin Receptor Antagonist and a PDE5 Inhibitor
| |
11-27-2009
|
Substituted PDE5 inhibitors
| |
9-4-2009
|
Uses of 2-Phenyl-Substituted Imidazotriazinone Derivatives for Treating Pulmonary Hypertension
| |
8-28-2009
|
Roflumilast for the Treatment of Pulmonary Hypertension
| |
8-7-2009
|
Use of Phosphodiesterase Inhibitor as a Component of Implantable Medical Devices
| |
6-26-2009
|
Method for healing a wound using a phosphodiesterase type five inhibitor
| |
3-20-2009
|
Pde5 inhibitor compositions and methods for immunotherapy
| |
3-6-2009
|
Pde5 inhibitor compositions and methods for treating cardiac indications
| |
10-31-2008
|
Formulations with Controlled Release of Active Ingredient
| |
8-15-2008
|
HIGHLY SELECTIVE and LONG-ACTING PDE5 MODULATORS
|
8-8-2008
|
Formulations With Controlled Release Of Active Ingredient
| |
4-11-2008
|
Use of 2-alkoxyphenyl-substituted imidazotriazinones
| |
1-2-2008
|
2-phenyl substituted imidazotriazinones as phosphodiesterase inhibitors, for treatment of hypertension
| |
12-28-2007
|
Novel Uses of 2-Phenyl-Substituted Imidazotriazinone Derivatives
| |
10-3-2007
|
Use of 2-alkoxyphenyl-substituted imidazotriazinones
| |
11-24-2006
|
Methods for synthesizing imidazotriazinones
| |
10-18-2006
|
2-Phenyl substituted imidazotriazinones as phosphodiesterase inhibitors
| |
2-15-2006
|
Process for the preparation of sulphonamide-substituted imidazotriazinones
| |
8-17-2005
|
Use of 2-alkoxyphenol-substituted imidazotriazinones
| |
5-11-2005
|
2-phenyl substituted imidazotriazinones as phosphodiesterase inhibitors
|
1-21-2005
|
Process for the preparation of sulphonamide-substituted imidazotriazinones
| |
8-18-2004
|
Process for the preparation of sulphonamide-substituted imidazotriazinones
| |
8-6-2004
|
Novel use of 2-phenyl-substituted imidazotriazinones
| |
7-32-2003
|
Daily treatment for erectile dysfunction using a PDE5 inhibitor
| |
5-21-2003
|
2-phenyl substituted imidatriazinones as phosphodiesterase inhibitors
| |
3-27-2002
|
2-phenyl substituted imidazotriazinones as phosphodiesterase inhibitors
| |
12-21-2001
|
Daily treatment for erectile dysfunction using a PDE5 inhibitor
| |
5-21-1999
|
2-PHENYL SUBSTITUTED IMIDAZOTRIAZINONES AS PHOSPHODIESTERASE INHIBITORS
|
..........................................................................
8. SILDENAFIL

1-[4-ethoxy-3-(6,7-dihydro-1-methyl-
7-oxo-3-propyl-1H-pyrazolo[4,3-d]pyrimidin-5-yl)
phenylsulfonyl]-4-methylpiperazine
CAS NO 139755-83-2
Sildenafil citrate, sold as Viagra, Revatio and under various other trade names, is a drug used to treat erectile dysfunction and pulmonary arterial hypertension (PAH). It was originally developed by British scientists and then brought to market by the US-based pharmaceutical company Pfizer.[1]It acts by inhibiting cGMP-specific phosphodiesterase type 5 (PDE5), an enzyme that promotes degradation of cGMP, which regulates blood flow in the penis. Since becoming available in 1998, sildenafil has been the prime treatment for erectile dysfunction; its primary competitors on the market are tadalafil (Cialis) and vardenafil (Levitra)
Erectile dysfunction (or ED), also called male impotence, describes a mans inability to achieve and maintain an erection sufficient for mutually satisfactory sexual intercourse with his partner. By itself, ED is not a disease but more of a signal that something else may be a problem.
Viagra works best if taken 30-60 minutes before sexual activity. Only 1 tablet should be taken per day. It should be taken on an empty stomach. Increasing the dosage beyond the recommended amounts will not improve the response and will only result in greater side effects.
Sildenafil is a vasoactive agent used to treat erectile dysfunction and reduce
symptoms in patients with pulmonary arterial hypertension (PAH).
symptoms in patients with pulmonary arterial hypertension (PAH).
Sildenafil elevates levels of the second messenger, cGMP, by inhibiting its breakdown
via phosphodiesterase type 5 (PDE5). PDE5 is found in particularly high
concentrations in the corpus cavernosum, erectile tissue of the penis.
via phosphodiesterase type 5 (PDE5). PDE5 is found in particularly high
concentrations in the corpus cavernosum, erectile tissue of the penis.
It is also found in the retina and vascular endothelium.
Increased cGMP results in vasodilation which facilitates generation and
maintenance of an erection. The vasodilatory effects of sildenafil also help reduce
symptoms of PAH.
Sildenafil citrate, sold as Viagra, Revatio and under various other trade names,
is adrug used to treat erectile dysfunction and pulmonary arterial
hypertension (PAH).
It was originally developed by British scientists and then brought to market by the
US-basedpharmaceutical company Pfizer.
maintenance of an erection. The vasodilatory effects of sildenafil also help reduce
symptoms of PAH.
Sildenafil citrate, sold as Viagra, Revatio and under various other trade names,
is adrug used to treat erectile dysfunction and pulmonary arterial
hypertension (PAH).
It was originally developed by British scientists and then brought to market by the
US-basedpharmaceutical company Pfizer.
It acts by inhibiting cGMP-specific phosphodiesterase type 5, an enzyme that promotes degradation of cGMP, which regulates blood flow in thepenis.
Since becoming available in 1998, sildenafil has been the prime treatment for erectile dysfunction; its primary competitors on the market are tadalafil (Cialis) and vardenafil (Levitra).
VIAGRA® (sildenafil citrate) , an oral therapy for erectile dysfunction, is the citrate salt of sildenafil, a selective inhibitor of cyclic guanosine monophosphate (cGMP)-specific phosphodiesterase type 5 (PDE5).
Sildenafil citrate is designated chemically as 1-[[3-(6,7-dihydro-1-methyl-7-oxo-3-propyl-1H pyrazolo[4,3-d]pyrimidin-5-yl)-4-ethoxyphenyl]sulfonyl]-4-methylpiperazine citrate and has the following structural formula:
 |
Sildenafil citrate is a white to off-white crystalline powder with a solubility of 3.5 mg/mL in water and a molecular weight of 666.7. VIAGRA (sildenafil citrate) is formulated as blue, film-coated rounded-diamond-shaped tablets equivalent to 25 mg, 50 mg and 100 mg of sildenafil for oral administration. In addition to the active ingredient, sildenafil citrate, each tablet contains the following inactive ingredients: microcrystalline cellulose, anhydrous dibasic calcium phosphate, croscarmellose sodium, magnesium stearate, hypromellose, titanium dioxide, lactose, triacetin, and FD & C Blue #2 aluminum lake.
Target is cyclic guanosine monophosphate phosphodiesterase type 5 receptor.
Fluforma is a company which found the genome explorations and target identification
through affymetrix microarrays.
through affymetrix microarrays.
High throughput functional assays of Si Rna’s inhibit the expression of target,
chromatographic techniques, spectrophotometric methods, adsorptive
stripping voltametry.
chromatographic techniques, spectrophotometric methods, adsorptive
stripping voltametry.
Gingel et.al showed that a 50mg dose given daily for 28 days consecutively
improved errections in almost 90% of patients.
Structural validation techniques are NMR technique, Infrared spectrum, HPLC, Mass spectroscopy and Liquid chromatography.improved errections in almost 90% of patients.

The chemical name of sildenafil is 5-[2-ethoxy-5-(4-methylpiperazin-1-ylsulfonyl)phenyl]-1- methyl-3-propyl-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one and its formula is C22H30N6O4S. The melting point of sildenafil is 189-190oC. Its solubility is 3.5 mg/mL in water. | |||||||||||||||||||||||||||||||||||||
 | |||||||||||||||||||||||||||||||||||||
The 1H NMR data of sildenafil is given below. The abbreviations used are s for singlet, d for doublet, t for triplet and q for quartet. The chemical shifts are given in ppm (parts per million) and are followed by the number of Hydrogens the peaks account for:
| |||||||||||||||||||||||||||||||||||||
 Viagra aims at inhibiting the enzyme phosphodiesterase PDE5. It must therefore have a structure that is similar in some places to the substrate. However, there are many other constraints as there are several different types of PDE enzymes which are found in different parts of the body. Of the 7 types of PDE, three selectively hydrolyse cGMP relative to cAMP. PDE5 itself can be found in several parts of the body : the lungs, platelets and various forms of smooth muscle. Selectivety was a very important factor in the research for an inhibitor of PDE5. The research on a molecule to inhibit PDE type 5 started with the molecule Zaprinast (1) which is shown on the right. The research established that derivatives of pyrazolo[4,3-d]pyrimidin-7-one (2) gave more potent cGMP PDE5 inhibition. The studies carried out compared many molecules by changing the substituents on them and comparing the inhibitory data of these molecules. To establish the selectivity of the compound, the compounds were also screened against the other widespread cGMP enzymes, PDE1 which was isolated from rat liver and PDE3 which was isolated from rat platelet. Further studies showed that sildenafil was the best PDE5 inhibitor. The different functional groups on the molecule were established by comparing the affinities of the molecules to the different PDE enzymes and establishing the selectivity of each compound made. Of course, high selectivity for PDE type 5 was looked for. Viagra mimics the guanosine base of cGMP and the extension of 3-substituent fills a space in the enzyme active site occupied by ribose. Substituents on the 5'-position of the phenyl ring reproduce the role of the phosphate bonding. To improve the solubility of the drug, polar substituents were added which gave compounds with a lower lipophilicity. This was found to also increase the enzyme affinity. Sidenafil gave an excellent combination of enzyme inhibitory potency, selectivity, solubility and in vivo characteristics. |  | ||||||||||||||||||||||||||||||||||||
It is interesting to compare the structure of sidenafil with that of cGMP (cyclic guanosine monophosphate), which is the molecule that usually interacts with PDE5. The drug that was being developed by the researchers had to have some similarity so that it could bind to the active site of the enzyme, PDE5. The 2 dimensional structure of cGMP is given above the 3 dimensional picture, however the latter can be used to view the molecule in 2D as well, with any orientation of the molecule. The first figure also shows that cGMP and pyrazolo[4,3-d]pyrimidin-7-one, which is one of the parent molecules of sildenafil, have a similar size, shape and dipole moment. These were characteristics that were looked for in a molecule that would inhibit PDE5. The following molecule is cGMP :  | |||||||||||||||||||||||||||||||||||||
Sildenafil is the active ingredient in Viagra ™ and it is chemically designated as 5-[2- ethoxy-5 -(4-methyl)piperazine- 1 -yl-sulfonyl)phenyl]- 1 -methyl-3-n-propyl- 1 ,6-dihydro- 7H-pyrazolo[4,3-d]pyrimidin-7-one, and the following chemical structure V:

V
Sildenafil is originally disclosed in US Patent No. 5,250,534 and it has been found to be particularly useful in the treatment of inter-alia male erectile dysfunction. Multi step syntheses for the production of Sildenafil are disclosed in US'534 with a yield of 27%.
An improved process for its preparation is described in a later application i.e. US Patent No. 5,955,611 which consisting the preparation of 5-chlorosulfonyl-2-ethoxybenzoic acid and converting it into 2-ethoxy-5-(4-methylpiperazin-l-ylsulfonayl)benzoic acid. The acid is then condensed with 4-amino-l-methyl-3-n-propylpyrazole-5-carboxyamide in the presence of N,N'-carbonyldiimidazole, and the resulting 4-[2-ethoxy-5-(4- methylpiperzin-l-ylsulfonyl)benzamido]-l-methyl-3-n-propylpyrazolo-5-carboxyamide is cyclized in an alkaline, neutral or acid solution to yield Sildenafil with about 47% yield.
Patent application number WO 2001/22918 describes the process for the preparation of Sildenafil wherein the process involves the reaction step between 2-ethoxy-5-(4-methyl- piperazine- 1 -sulfonyl)benzaldehyde and 4-amino- 1 -methyl-S-n-propyl-pyrazole-S- carboxamide to form an intermediate which is further cyclised to form Sildenafil.
Patents/ patent applications US6204383, US20030069422, US20030144530, US 20040106796, US 20040110948, EP 0812845, EP 1002798, EP 1077214, WO 01/19827, WO 04/31134 also discloses several processes for the preparation of Sildenafil citrate.
Sildenafil citrate is a white to off-white crystalline powder with a solubility of 3.5 mg/mL in water and a molecular weight of 666.7. Sildenafil citrate has most recently been utilized as the basis for an oral therapy for erectile dysfunction and has been marketed by Pfizer Labs under the trademark Viagra®. Publications relating to benign visual side-effects (e.g., blue-shift in vision, light-sensitivity, and blurring noted to occur in some patients) of sildenafil prompted the FDA to insist on product insert warnings.
Sildenafil citrate is a white to off-white crystalline powder with a solubility of 3.5 mg/mL in water and a molecular weight of 666.7. Sildenafil citrate has most recently been utilized as the basis for an oral therapy for erectile dysfunction and has been marketed by Pfizer Labs under the trademark Viagra®. Publications relating to benign visual side-effects (e.g., blue-shift in vision, light-sensitivity, and blurring noted to occur in some patients) of sildenafil prompted the FDA to insist on product insert warnings.
Sexual dysfunction
The primary indication of sildenafil is treatment of erectile dysfunction (inability to sustain a satisfactory erection to complete intercourse). Its use is now standard treatment for erectile dysfunction in all settings, including diabetes.[2]
People on antidepressants may experience sexual dysfunction, either as a result of their illness or as a result of their treatment. A 2003 study showed that sildenafil improved sexual function in men in this situation.[3] Following up reports from 1999,[4] the same researchers found that sildenafil improved sexual function in female patients on antidepressants as well.[5]
Pulmonary hypertension
As well as erectile dysfunction, sildenafil citrate is also effective in the rare disease pulmonary arterial hypertension (PAH). It relaxes the arterial wall, leading to decreased pulmonary arterial resistance and pressure. This, in turn, reduces the workload of the right ventricle of the heart and improves symptoms of right-sided heart failure. Because PDE5 is primarily distributed within the arterial wall smooth muscle of the lungs and penis, sildenafil acts selectively in both these areas without inducing vasodilation in other areas of the body. Pfizer submitted an additional registration for sildenafil to the United States Food and Drug Administration (FDA), and sildenafil was approved for this indication in June 2005. The preparation is named Revatio, to avoid confusion with Viagra, and the 20 milligram tablets are white and round. Sildenafil joins bosentan and prostacyclin-based therapies for this condition.[6]

|



SCHEME2
Example 1
Preparation of 2- hydroxy-5-(4 methyl)-l-piperazinyl sulphonyl) benzoic acid Step-1: Preparation of 5-Chlorosulfonyl-2-hydroxy benzoic acid
To the chilled chlorosulfonic acid (1012 g), salicylic acid (200 g) was added at 0-50C over a period of 1 hour 40 min. The temperature of the reaction mixture was maintained at 20-250C for 2 hrs. Then thionyl chloride (172.4 g) was added over a period of 15 min and maintained for 12 hrs. The product formed was poured onto ice and maintained for lhr. The product was filtered and washed with DM water to get 5-Chlorosulfonyl-2- hydroxy benzoic acid.
Step-2: Preparation of 2-hydroxy-5-(4-methyI)-l-piperazinylsulphonyl)benzoic acid
5-Chlorosulfonyl-2-hydroxy benzoic acid (40Og) obtained in step 1 was dissolved in acetone (1200 ml) and cooled to 5-100C. To this clear solution N-methyl piperazine (254 g) was added and maintained for 2 hrs. The product formed was filtered, washed with water and purified in methanol to get 308 g of the titled compound.
NMR Data:
1H-NMR (300 MHz in DMSO-d6): δ 2.78 (3H, s), 3.17 (8H, brs), 6.85(1H, d, J = 8.7),
7.52 - 7.56 (IH, dd, J=8.7, 2.7), 7.95 (IH, d, J = 2.7)
13C-NMR (75 MHz in DMSO-d6): δ 41.98, 43.36, 51.60, 117.58, 118.33, 119.46,
130.28, 132.01, 167.63, 170.35.
Melting point: 268-2720C
Purity by HPLC: 99.4% Example 2
Preparation of 4-[2-hydroxy-5-(4-methyI-l-piperazinyIsulphonyl)benzamido]-l- methyl-3-n-propyl-lH-pyrazole-5-carboxamide
2-Hydroxy-5-(4-methyl-l-piperazinylsulphonyl)benzoic acid (10Og) was dissolved in dichloromethane (500 ml) and triethylamine (50 ml) followed by distillation to get residual mass. The residual mass was dissolved in dichloromethane (1500ml) followed by the addition of 1,3-dicyclohexylcarbodiimide (75.6 g) and 1-hydroxybenzotriazole (45g). The reaction mixture was stirred at 27-280C and then 4-amino-l-methyl-3-n-propyl- pyrazole-5-carboxamide (60.6 g) was added. The reaction mixture was heated to reflux temperature and maintained for 3 hours. Filtered the undissolved material at hot and washed the cake with dichloromethane (200ml). The filtrate was distilled out completely to get residue. Dissloved the residue in methanol (300ml) at 4O0C and then cooled the mass to 27-280C and stirred overnight. Further, cooled the mass to 5-70C and stirred for lhr. Filtered the product and washed the cake with chilled methanol (100ml) and dried to get 130 g of title compound.
NMR Data:
1H-NMR (300 MHz in DMSO-d6): δ 0.87 (3H, t, J = 7.5), 1.53-1.60 (2H, m), 2.39- 2.46(5H, m), 2.72 (4H, brs), 2.96 (4H, brs), 3.17 (3H, s), 3.91 (3H, s), 6.93 (H, d, J = 8.7), 7.57-7.61 (H, dd, J=8.7 & 2.1), NH2-(2H, brs, J =7.69 & 7.72), 8.15 (IH, d, J=2.1) 11.5 (OH, br).
13C-NMR (75 MHz in DMSO-(I6): 613.80, 21.37, 27.45, 44.05, 44.75, 48.60, 52.87, 116.37, 118.06, 119.67, 120.03, 130.64, 132.17, 132.38, 146.16, 160.83, 166.33, 166.89.
Purity by HPLC: 97.5%
Example 3
Preparation of 5-[2-hydroxy-5-(4-methylpiperazinyl-l-yl-sulphonyl)phenyl]-l- methyl -3-n- propyl-l,6-dihydro-7H-pyrazolo-[4,3-d]pyrimidin-7-one Sodium hydroxide (34 g) was added into diethylene glycol (780ml) and then heated to 110-1150C. 4-[2-hydroxy-5-(4-methyl-l-piperazinylsulphonyl)benzamido)-l-methyl-3-n- propyl-lH-pyrazole-5-carboxamide (130 g) obtained from example 2 was added to the above reaction mixture. The reaction mixture was maintained at 125-13O0C for 4-6 hrs. The reaction mixture was cooled to room temperature and then DM water (1300ml) was added slowly over 20min at 250C and maintained at this temperature for 1 hour. Filtered the mass and filtrate pH was adjusted to 6.5-7.5 with dilute hydrochloric acid at room temperature and stirred at room temperature for 2-3hrs. Product was filtered and slurried the cake with excess DM Water followed by purification in methanol to get 91 g of titled compound.
NMR Data:
1H-NMR (300 MHz in DMSO-d6): δ 0.96 (3H, t, J=7.2), 1.71-1.83 (2H, m), 2.41 (3H, s), 2.78-2.83 (6H, m), 2.99 (4H, brs), 4.15 (3H3 s), 6.93 (IH, d, J=8.7), 7.54-7.57 (IH, dd, J=8.7, 2.1), 8.47 (lH, d, J=2.1).
13C-NMR (75MHz in DMSO-d6): 513.84, 21.52, 27.20, 37.80, 43.94, 44.72,- 52.80, 115.97, 119.82, 120.19, 124.48, 128.71, 131.13, 136.46, 143.82, 151.26, 154.05, 167.24.
Purity by HPLC: 97.8%
Example 4
Preparation of 5-[2-ethoxycarbonyloxy-5-(4-methylpiperazin-l-yl-sulfonyI)phenyl]- l-methyI-3n-propyI-l,6-dihydro-7H-pyrazolo-[4,3-d]pyrimidin-7-one
5-[2-hydroxy-5-(4-methylpiperazinyl-l -yl-sulphonyl)phenyl]- 1 -methyl-3 -n- propyl- 1 ,6- dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one (90 g) obtained from example 3 was dissolved in dichloromethane (360 ml) and added triethyl amine (41 ml) at room temperature and stirred for 10 min. The reaction mixture was cooled to 0-50C and followed by the addition of ethyl chloro formate (24ml) over 30 min under nitrogen atmosphere. The temperature of the reaction was raised slowly to 28-3O0C and maintained for 24 hrs. The reaction mixture was cooled to 0-50C and kept it for 1 hr. The product formed was filtered, washed with dichloromethane, dried and purified from methanol (270ml) to obtain 81 g of the title compound.
NMR Data:
1H-NMR (300 MHz in DMSO-d6): δ 0.92 (3H, t, J=7.2), 1.17 (3H, t, J=7.2), 1.68-1.75 (2H, m), 2.16 (3H, s), 3.99 (4H, br), 2.73 (2H, t, J=7.0), 4.12-4.19 (2H, t, J=6.9), 4.15 (3H, s), 7.71 (IH, d, J = 8.7), 7.93-7.97 (IH, dd, J=8.7 & 2.1), 8.01 (IH, d, J=2.0)
13C-NMR (75 MHz in DMSO-d6): 513.47, 13.80, 21.57, 27.03, 37.90, 45.72, 53.49, 65.12, 124.51, 127.65, 130.14, 130.61, 132.82, 137.30, 144.96, 146.51, 151.38, 151.66, 154.36.
Purity by HPLC: 98.6%
Example 5
Preparation of 5-[2-ethoxy-5-(4-methyl piperazine-l-ylsulfonyl)phenyl]-l-methyl-3- n-propyl-1 ,6-dihydro-7H-pyrazolo [4,3-d] pyrimidin-7-one (Sildenafil base)
5-[2-Ethoxycarbonyloxy-5-(4-methylpiperazin-l-yl-sulfonyl)phenyl]-l-methyl-3-n- propyl-l,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one (50g) was dissolved in ethanol (750ml) in an autoclave and then added dicyclohexylcarbodimide (29.8g). The reaction temperature was raised to HO0C with internal pressure of 1.8-4.0 kg/cm and maintained for 6 hours followed by cooling to room temperature. The solvent was distilled off to get the crude Sildenafil base. The base thus obtained was dissolved in dichloromethane (380ml), filtered and filtrate was distilled out completely to get solid material, which is again dissolved in a mixture dichloromethane and isopropyl ether. The crude obtained was recrystallized from ethanol (260ml) to obtain 17.4gm of pure Sildenafil base.
Purity by HPLC: 99.77% Example 6
Preparation of 5-[2-Ethoxy-5-(4-methylpiperazine-l-yI-sulfonyl)phenyl]-l-methyl- 3-n-propyl-l,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one citrate (Sildenafil Citrate)
Sildenafil base (50 g) was dissolved in acetone (850 ml) at 550C and then slowly added citric acid solution (20 g in 100 ml acetone) over 45 min and maintain the reaction mixture for about 30 min. The reaction mixture was cooled, filtered and dried to get 65 g of Sildenafil citrate.
Purity by HPLC: 99.85%
Chemical synthesis
The preparation steps for synthesis of sildenafil are as follows:[45]
- Methylation of 3-propylpyrazole-5-carboxylic acid ethyl ester with hot dimethyl sulfate
- Hydrolysis with aqueous NaOH to free acid
- Nitration with oleum/fuming nitric acid
- Carboxamide formation with refluxing thionyl chloride/NH4OH
- Reduction of nitro group to amino
- Acylation with 2-ethoxybenzoyl chloride
- Cyclization
- Sulfonation to the chlorosulfonyl derivative
- Condensation with 1-methylpiperazine.
SYNTHESIS
...................................................
...........................................................................
SYNTHESIS
......................................................................................
.................................
PRECURSORS
.........................................
SYNTHESIS
Patent issues and expirations
European Union
Pfizer's patent on sildenafil citrate expired in some member countries of the EU, Austria, Denmark, France, Germany, Ireland, Italy, The Netherlands, Spain, Sweden, United Kingdom as well as Switzerland on 21 June 2013.[60][61][62] A UK patent held by Pfizer on the use of PDE5 inhibitors (see below) as treatment of impotence was invalidated in 2000 because of obviousness; this decision was upheld on appeal in 2002.[63][64]
United States
In 1992 Pfizer filed a patent covering the substance sildenafil and its use to treat cardiovascular diseases.[65] This patent was published in 1993 and expired in 2012. In 1994 Pfizer filed a patent covering the use of sildenafil to treat erectile dysfunction.[66] This patent was published in 2002 and will expire in 2019. Teva sued to have the latter patent invalidated, but Pfizer prevailed in an August 2011 federal district court case.[67]
The patent on Revatio (indicated for pulmonary arterial hypertension rather than erectile dysfunction) expired in late 2012. Generic versions of this low-dose form of sildenafil have been available in the U.S. from a number of manufacturers including Greenstone, Mylan and Watson, since early 2013.[68] There is no legal barrier to doctors prescribing this form of sildenafil "off label" for erectile dysfunction, although the dosage typically required for treating ED requires patients to take multiple pills.
Canada
In Canada, Pfizer's patent 2,324,324 for Revatio (sildenafil used to treat pulmonary hypertension) was found invalid by the Federal Court in June 2010, on an application by Ratiopharm Inc.[69][70]
On November 8, 2012 the Supreme Court of Canada ruled that Pfizer's patent 2,163,446 on Viagra was invalid from the beginning because the company did not provide full disclosure in its application. The decision, Teva Canada Ltd. v. Pfizer Canada Inc., pointed to section 27(3)(b) of The Patent Act which requires that disclosure must include sufficient information "to enable any person skilled in the art or science to which it pertains" to produce it. It added further: "As a matter of policy and sound statutory interpretation, patentees cannot be allowed to 'game' the system in this way. This, in my view, is the key issue in this appeal."[71]
Teva Canada launched Novo-Sildenafil, a generic version of Viagra, on the day the Supreme Court of Canada released its decision.[72][73][74] To remain competitive, Pfizer then reduced the price of Viagra in Canada.[75] However, on November 9, 2012, Pfizer filed a motion for a re-hearing of the appeal in the Supreme Court of Canada,[76] on the grounds that the court accidentally exceeded its jurisdiction by voiding the patent.[77] Finally, on April 22, 2013, The Supreme Court of Canada invalidated Pfizer's patent altogether.[78]
India
Manufacture and sale of sildenafil citrate drugs known as "generic viagra" is common in India, where Pfizer's patent claim does not apply. Trade names include Kamagra (Ajanta Pharma), Silagra (Cipla), Edegra (Sun Pharmaceutical), Penegra (Zydus Cadila), and Zenegra (Alkem Laboratories).
China
Manufacture and sale of sildenafil citrate drugs known as "generic viagra" is common in China, where Pfizer's patent claim is not widely enforced.
Other countries
Egypt approved Viagra for sale in 2002, but soon afterwards allowed local companies to produce generic versions of the drug, citing the interests of poor people who would not be able to afford Pfizer's price.[79]
Pfizer's patent on sildenafil citrate expired in Brazil in 2010.[80]
References
- ^ a b Boolell M, Allen MJ, Ballard SA, Gepi-Attee S, Muirhead GJ, Naylor AM, Osterloh IH, Gingell C (June 1996). "Sildenafil: an orally active type 5 cyclic GMP-specific phosphodiesterase inhibitor for the treatment of penile erectile dysfunction". Int. J. Impot. Res. 8 (2): 47–52.PMID 8858389.
- ^ Vardi M, Nini A (2007). "Phosphodiesterase inhibitors for erectile dysfunction in patients with diabetes mellitus". In Vardi, Moshe. Cochrane Database Syst Rev (1): CD002187.doi:10.1002/14651858.CD002187.pub3. PMID 17253475.
- ^ Nurnberg HG, Hensley PL, Gelenberg AJ, Fava M, Lauriello J, Paine S (January 2003)."Treatment of antidepressant-associated sexual dysfunction with sildenafil: a randomized controlled trial". JAMA 289 (1): 56–64. doi:10.1001/jama.289.1.56. PMID 12503977.
- ^ Nurnberg HG, Hensley PL, Lauriello J, Parker LM, Keith SJ (August 1999). "Sildenafil for women patients with antidepressant-induced sexual dysfunction". Psychiatr Serv 50 (8): 1076–8. PMID 10445658.
- ^ Nurnberg HG, Hensley PL, Heiman JR, Croft HA, Debattista C, Paine S (2008). "Sildenafil Treatment of Women With Antidepressant-Associated Sexual Dysfunction". JAMA 300 (4): 395–404. doi:10.1001/jama.300.4.395. PMID 18647982.
- ^ "FDA Approves Pfizer's Revatio as Treatment for Pulmonary Arterial Hypertension" (Press release). Pfizer. June 6, 2005. Archived from the original on August 28, 2005.
- ^ Richalet JP, Gratadour P, Robach P, et al. (2005). "Sildenafil inhibits altitude-induced hypoxemia and pulmonary hypertension". Am. J. Respir. Crit. Care Med. 171 (3): 275–81.doi:10.1164/rccm.200406-804OC. PMID 15516532.
- ^ Perimenis P (2005). "Sildenafil for the treatment of altitude-induced hypoxaemia". Expert Opin Pharmacother 6 (5): 835–7. doi:10.1517/14656566.6.5.835. PMID 15934909.
- ^ Fagenholz PJ, Gutman JA, Murray AF, Harris NS (2007). "Treatment of high altitude pulmonary edema at 4240 m in Nepal". High Alt. Med. Biol. 8 (2): 139–46.doi:10.1089/ham.2007.3055. PMID 17584008.
- ^ "Pill Identifier". Drugs.com. Retrieved 2009-02-10. "This site is intended for viewing by the USA audience only. If you are in another country, local laws may not permit access to the medical information contained in this site."
- ^ a b c "Viagra Prescribing Information" (PDF). Pfizer. October 2007. Archived from the original on 14 November 2012. Retrieved 14 November 2012.
- ^ "Viagra and vision". VisionWeb. 29 October 2001. Retrieved 2009-02-10.
- ^ "FDA Updates Labeling for Viagra, Cialis and Levitra for Rare Post-Marketing Reports of Eye Problems". United States Food and Drug Administration. 8 July 2005. Archived from the original on February 23, 2008. Retrieved 2009-02-10.
- ^ Pomeranz HD, Bhavsar AR (March 2005). "Nonarteritic ischemic optic neuropathy developing soon after use of sildenafil (viagra): a report of seven new cases". J Neuroophthalmol 25 (1): 9–13. doi:10.1097/00041327-200503000-00003. PMID 15756125.
- ^ Egan R, Pomeranz H (February 2000). "Sildenafil (Viagra) associated anterior ischemic optic neuropathy". Arch. Ophthalmol. 118 (2): 291–2. PMID 10676804.
- ^ Pomeranz HD, Smith KH, Hart WM, Egan RA (March 2002). "Sildenafil-associated nonarteritic anterior ischemic optic neuropathy". Ophthalmology 109 (3): 584–7. doi:10.1016/S0161-6420(01)00976-9. PMID 11874765.
- ^ Cunningham AV, Smith KH (March 2001). "Anterior ischemic optic neuropathy associated with viagra". J Neuroophthalmol 21 (1): 22–5. doi:10.1097/00041327-200103000-00006.PMID 11315976.
- ^ Boshier A, Pambakian N, Shakir SA (September 2002). "A case of nonarteritic ischemic optic neuropathy (NAION) in a male patient taking sildenafil". Int J Clin Pharmacol Ther 40 (9): 422–3. PMID 12358159.
- ^ Akash R, Hrishikesh D, Amith P, Sabah S (August 2005). "Case report: association of combined nonarteritic anterior ischemic optic neuropathy (NAION) and obstruction of cilioretinal artery with overdose of Viagra". J Ocul Pharmacol Ther 21 (4): 315–7.doi:10.1089/jop.2005.21.315. PMID 16117695.
- ^ "FDA Announces Revisions to Labels for Cialis, Levitra and Viagra". United States Food and Drug Administration. 18 October 2007. Archived from the original on 11 July 2007. Retrieved 10 February 2009.
- ^ "Viagra (sildenafil citrate) tablets". page 29: Pzifer. October 2007. Retrieved 2009-10-25.
- ^ Kloner RA (2005). "Pharmacology and drug interaction effects of the phosphodiesterase 5 inhibitors: focus on alpha-blocker interactions". Am J Cardiol 96 (12B): 42M–46M.doi:10.1016/j.amjcard.2005.07.011. PMID 16387566.
- ^ Cheitlin MD, Hutter AM Jr, Brindis RG, Ganz P, Kaul S, Russell RO Jr, Zusman RM (1999). "ACC/AHA expert consensus document. Use of sildenafil (Viagra) in patients with cardiovascular disease. American College of Cardiology/American Heart Association". Journal of the American College of Cardiology 33 (1): 273–82. doi:10.1016/S0735-1097(98)00656-1.PMID 9935041.
- ^ Peterson K (2001-03-21). "Young men add Viagra to their drug arsenal". USAToday.
- ^ a b c d e Smith KM, Romanelli F (2005). "Recreational use and misuse of phosphodiesterase 5 inhibitors". J Am Pharm Assoc (2003) 45 (1): 63–72; quiz 73–5.doi:10.1331/1544345052843165. PMID 15730119.
- ^ Sildenafil Will Not Affect Libido - Fact!
- ^ a b Mondaini N, Ponchietti R, Muir GH, Montorsi F, Di Loro F, Lombardi G, Rizzo M (June 2003). "Sildenafil does not improve sexual function in men without erectile dysfunction but does reduce the postorgasmic refractory time". Int. J. Impot. Res. 15 (3): 225–8.doi:10.1038/sj.ijir.3901005. PMID 12904810.
- ^ McCambridge J, Mitcheson L, Hunt N, Winstock A (March 2006). "The rise of Viagra among British illicit drug users: 5-year survey data". Drug Alcohol Rev 25 (2): 111–3.doi:10.1080/09595230500537167. PMID 16627299.
- ^ Eloi-Stiven ML, Channaveeraiah N, Christos PJ, Finkel M, Reddy R (November 2007). "Does marijuana use play a role in the recreational use of sildenafil?". J Fam Pract 56 (11): E1–4.PMID 17976333.
- ^ "The 2007 Ig Nobel Prize Winners". Improbable Research. 4 October 2007. Retrieved 2009-02-10.
- ^ Agostino PV, Plano SA, Golombek DA (June 2007). "Sildenafil accelerates reentrainment of circadian rhythms after advancing light schedules". Proc. Natl. Acad. Sci. U.S.A. 104 (23): 9834–9. doi:10.1073/pnas.0703388104. PMC 1887561. PMID 17519328.
- ^ Teri Thompson, Christian Red, Michael O'Keefffe, and Nathaniel Vinton (10 June 2008)."Source: Roger Clemens, host of athletes pop Viagra to help onfield performance". Daily News (Daily News). Retrieved 2009-02-10.
- ^ Busbee J (2012-11-28). "Bears’ Brandon Marshall says some NFL players use Viagra … ON THE FIELD". Yahoo! Sports. Retrieved 2012-11-28.
- ^ Siegel-Itzkovich J (July 1999). "In brief: Viagra makes flowers stand up straight". BMJ 319(7205): 274B. doi:10.1136/bmj.319.7205.274a. PMC 1126921. PMID 10426724.
- ^ "Prolongation of the shelf life of fruits and flowers". Biology Department, University of Hamburg. Retrieved 28 November 2012.
- ^ Oh SS, Zou P, Low MY, Koh HL. Detection of sildenafil analogues in herbal products for erectile dysfunction (2006). "Detection of sildenafil analogues in herbal products for erectile dysfunction". Journal of Toxicology and Environmental Health Part A 69 (21): 1951–1958.doi:10.1080/15287390600751355. PMID 16982533.
- ^ Venhuis BJ, Blok-Tip L, de Kaste D (2008). "Designer drugs in herbal aphrodisiacs".Forensic Science International 177 (2–3): 25–27. doi:10.1016/j.forsciint.2007.11.007.PMID 18178354.
- ^ FDA letter to Libidus distributor
- ^ FDA Warns Consumers About Dangerous Ingredients in "Dietary Supplements" Promoted for Sexual Enhancement
- ^ Hidden Risks of Erectile Dysfunction "Treatments" Sold Online
- ^ R. Baselt, Disposition of Toxic Drugs and Chemicals in Man, 9th edition, Biomedical Publications, Seal Beach, CA, 2011, pp. 1552–1553.http://www.biomedicalpublications.com/dt9.pdf
- ^ Sung, B. J.; Hwang, K.; Jeon, Y.; Lee, J. I.; Heo, Y. S.; Kim, J.; Moon, J.; Yoon, J.; Hyun, Y. L.; Kim, E.; Eum, S.; Park, S. Y.; Lee, J. O.; Lee, T.; Ro, S.; Cho, J. (2003). "Structure of the catalytic domain of human phosphodiesterase 5 with bound drug molecules". Nature 425 (6953): 98–102. doi:10.1038/nature01914. PMID 12955149.
- ^ a b Webb, D.J.; Freestone, S.; Allen, M.J.; Muirhead, G.J. (March 4, 1999). "Sildenafil citrate and blood-pressure-lowering drugs: results of drug interaction studies with an organic nitrate and a calcium antagonist". Am. J. Cardiol. 83 (5A): 21C–28C. doi:10.1016/S0002-9149(99)00044-2. PMID 10078539.
- ^ "Viagra Clinical Pharmacology". RxList.com. 2008. Retrieved 2008-08-20.
- ^ Dunn PJ (2005). "Synthesis of Commercial Phosphodiesterase(V) Inhibitors". Org Process Res Dev 2005 (1): 88–97. doi:10.1021/op040019c.
- ^ "Research". ABM. Abertawe Bro Morgannwg University Health Board. 4 July 2008. Archived from the original on 26 September 2008. Retrieved 6 August 2008. "Our clinicians regularly offer patients the opportunity to take part in trials of new drugs and treatments. Morriston Hospital in Swansea, was the first in the world to trial Viagra!"
- ^ Terrett NK, Bell AS, Brown D, Elllis P (1996). "Sildenafil (Viagra), a potent and selective inhibitor of Type 5 cGMP phosphodiesterase with utility for the treatment of male erectile dysfunction". Bioorg Med Chem Lett 6 (15): 1819–1824. doi:10.1016/0960-894X(96)00323-X.
- ^ Kling J (1998). "From hypertension to angina to Viagra". Mod. Drug Discov. 1: 31–38.ISSN 1532-4486. OCLC 41105083.
- ^ Viagra sales peak at $1,934m in 2008
- ^ Bellis M. "Viagra, the patenting of an aphrodisiac". About.com. Retrieved 2009-02-10.
- ^ Ciment, J (1999). "Missouri fines internet pharmacy". BMJ (British Medical Journal) 319(7221): 1324. doi:10.1136/bmj.319.7221.1324g. PMC 1174637. PMID 10567131.
- ^ Devine, Amy (September 29, 2008). "Chemists plan to sell Viagra on the internet". Daily Record. Retrieved 2012-04-30.
- ^ "Urban Dictionary: Vitamin V". Urban Dictionary. January 16, 2009. Retrieved 2013-12-27.
- ^ Keith A (2000). "The economics of Viagra". Health Aff (Millwood) 19 (2): 147–57.doi:10.1377/hlthaff.19.2.147. PMID 10718028.
- ^ McGuire S (2007-01-01). "Cialis gaining market share worldwide". Medical Marketing & Media. Haymarket Media. Archived from the original on 2009-01-03. Retrieved 2009-02-10.
- ^ Mullin, Rick (June 20, 2005). "Viagra". Chemical & Engineering News 83 (25). Retrieved 2008-08-20.
- ^ Berenson, Alex (December 4, 2005). "Sales of Impotence Drugs Fall, Defying Expectations". The New York Times. Retrieved 2013-12-27.
- ^ "Over-the-counter Viagra piloted". BBC News. BBC News. 11 February 2007. Retrieved 2009-02-10.
- ^ "Pfizer to sell Viagra online, in first for Big Pharma: AP". CBS News. Retrieved 6 May 2013.
- ^ "Actavis Launches Generic Viagra in Europe as Patents Expire". Retrieved 2013-10-25.
- ^ Jim Edwards (October 21, 2009). "What Will Happen When Viagra Goes Generic?". AccessRx.com. Retrieved 2013-12-27.
- ^ "Is Viagra about to lose its pulling power in the UK?". The Guardian. Retrieved 13 June 2013.
- ^ Murray-, Rosie (23 January 2002). "Viagra ruling upsets Pfizer". London: Telegraph Media Group Limited. Archived from the original on 22 August 2009. Retrieved 10 February 2009.
- ^ "Pfizer Loses UK Battle on Viagra Patent". UroToday. Thomson Reuters. 17 June 2002. Archived from the original on 25 June 2007. Retrieved 10 February 2009.
- ^ U.S. Patent 5,250,534
- ^ U.S. Patent 6,469,012
- ^ "Pfizer Wins Viagra Patent Infringement Case Against Teva Pharmaceuticals". Bloomberg. August 15, 2011. Retrieved 2012-04-01.
- ^ "Pfizer's Revatio Goes Generic". Zacks Equity Research. November 15, 2012. Retrieved 2013-10-05.
- ^ "Revation patent ruled invalid for lack of sound prediction and obviousness". Canadian Technology & IP Law. Stikeman Elliott. 2010-06-18. Retrieved 2012-11-14.
- ^ "Pfizer Canada Inc. v. Ratiopharm Inc., 2010 FC 612". CanLII.
- ^ Teva Canada Ltd. v. Pfizer Canada Inc. 2012 SCC 60 at par. 80 (8 November 2012)
- ^ John Spears (2012-11-08). "Supreme Court ruling could lead to cheaper versions of Viagra". The [Toronto] Star. Retrieved 2012-11-14.
- ^ Ken Hanly (2012-11-08). "Canadian Supreme court rules Viagra patent invalid". Digital Journal. Retrieved 2012-11-14.
- ^ "Viagra patent tossed out by Supreme Court: Decision allows generic versions of drug to be produced". CBC News. 2012-11-08. Retrieved 2012-11-14.
- ^ "Pfizer Canada drops Viagra price after generic versions get Supreme Court green light".Financial Post. 2012-11-22. Retrieved 2013-12-27.
- ^ "SCC Case Information, Docket No. 33951". Retrieved 2012-11-14.
- ^ Kirk Makin (2012-11-15). "In rare move, Pfizer asks Supreme Court to reconsider ruling that killed Viagra patent". The Globe and Mail. Retrieved 2012-11-15.
- ^ Gowling Lafleur Henderson LLP, Hélène D'Iorio (2013-04-22). "The Supreme Court of Canada holds Pfizer’s Viagra patent invalid". Lexology. Retrieved 2013-12-27.
- ^ Allam, Abeer (October 4, 2002). "Seeking Investment, Egypt Tries Patent Laws". New York Times. Retrieved 2013-12-27.
- ^ Viagra patent expires in June, says Brazilian court
External links
Official Viagra Website- Official Viagra UK Website
- Official Revatio Website
- prescribing information for Viagra and prescribing information for Revatio from Pfizer
- FDA Information
- MedlinePLUS information, including side effects
- U.S. National Library of Medicine: Drug Information Portal – Sildenafil
- Viagra bound to proteins in the PDB
- Viagra at The Periodic Table of Videos (University of Nottingham)
9 TADALAFIL
Tadalafil (cialis)
Tadalafil
GF-196960, IC-351, Cialis
6R-trans)-6-(1,3-benzodioxol-5-yl)- 2,3,6,7,12,12a-hexahydro-2-methyl-pyrazino [1', 2':1,6] pyrido[3,4-b]indole-1,4-dione
Pyrazino[1',2':1,6]pyrido[3,4-b]indole-1,4-dione,6-(1,3-benzodioxol-5-yl)-2,3,6,7,12,12a-hexahydro-2-methyl-, (6R-trans)-; (6R,12aR)-6-(1,3-benzodioxol-5-yl)-2,3,6,7,12,12a-hexahydro-2-ethylpyrazino[1',2':1,6]pyrido[3,4-b]indole-1,4-dione; GF 196960; Adcirca;
171596-29-5 casno
Molecular Weight:
389.40
389.40
Molecular Formula:C22H19N3O4
GlaxoSmithKline (Originator), Lilly Icos (Marketer), Lilly (Licensee), Lilly Icos (Licensee)
Launched-2003
Tadalafil is currently marketed as Cialis. Cialis was developed by Eli Lilly as a treatment for impotence. In this capacity, it is reported that tadalafil functions by inhibiting the formation of cyclic guanosine monophosphate (cGMP)-specific phosphodiesterase type 5 (PDE5). The inhibition of PDE5 presumably lessens impotence by increasing the amount ot c(iMP, resulting in smooth muscle relaxation and increased blood flow.
Tadalafil is a PDE5 inhibitor marketed in pill form for treating erectile dysfunction (ED) under the name Cialis, and under the name Adcirca for the treatment of pulmonary arterial hypertension. In October 2011 the U.S. Food and Drug Administration (FDA) approved Cialis for treating the signs and symptoms of benign prostatic hyperplasia (BPH) as well as a combination of BPH and erectile dysfunction (ED) when the conditions coincide. It initially was developed by the biotechnology company ICOS, and then again developed and marketed world-wide by Lilly ICOS, LLC, the joint venture of ICOS Corporation and Eli Lilly and Company. Cialis tablets, in 2.5 mg, 5 mg, 10 mg, and 20 mg doses, are yellow, film-coated, and almond-shaped. The approved dose for pulmonary arterial hypertension is 40 mg (two 20-mg tablets) once daily.
Tadalafil can be prepared via a series of intermediates. One synthesis scheme is illustrated in Scheme 1: Scheme 1
U.S. Patent No. 5,859,006 describes the synthesis of the tadalafil intermediate (Compound III) from D-tryptophan methyl ester (Compound II) and piperonal (Compound I) using trifluoroacetic acid and dichloromethane, a halogenated solvent. Compound III is then reacted with chloroacetyl chloride (Compound IV) and chloroform, providing another intermediate of tadalafil (Compound V). WO 04/011463 describes a process of preparing tadalafil intermediates from D-tryptophan methyl ester HCl salt and piperonal by refluxing the reagents in isopropyl alcohol; the obtained intermediate is reacted with chloroacetyl chloride and THF, resulting in another intermediate of tadalafil.
Tadalafil is also manufactured and sold under the name of Tadacip by the Indian pharmaceutical company Cipla in doses of 10 mg and 20 mg.
On November 21, 2003 the FDA approved tadalafil (as Cialis) for sale in the United States as the third ED prescription drug pill (after sildenafil citrate(Viagra) and vardenafil (Levitra)). Like sildenafil and vardenafil, tadalafil is recommended as an ‘as needed’ medication. Cialis is the only one of the three that is also offered as a once-daily medication.
Moreover, tadalafil was approved in May 2009 in the United States for the treatment of pulmonary arterial hypertension and is under regulatory review in other regions for this condition. In late November 2008, Eli Lilly sold the exclusive rights to commercialize tadalafil for pulmonary arterial hypertension in the United States to United Therapeutics for an upfront payment of $150 million.
The FDA’s approval of Viagra (Sildenafil) on March 27, 1998 was a ground-breaking commercial event for the treatment of ED, with sales exceedingUS$1 billion. Subsequently, the FDA approved Levitra (vardenafil) on August 19, 2003, and Cialis (tadalafil) on November 21, 2003.
Cialis was discovered by Glaxo Wellcome (now GlaxoSmithKline) under a partnership between Glaxo and ICOS to develop new drugs that began in August 1991. [1][2] In 1993, the Bothell, Washington biotechnology company ICOS Corporation began studying compound IC351, a phosphodiesterase type 5 (PDE5) enzyme inhibitor. In 1994, Pfizer scientists discovered that sildenafil, which also inhibits the PDE5 enzyme, caused penile erection in men participating in a clinical study of a heart medicine. Although ICOS scientists were not testing compound IC351 for treating ED, they recognized its potential usefulness for treating that disorder. Soon, in 1994, ICOS received a patent for compound IC351 (structurally unlike sildenafil and vardenafil), and Phase 1 clinical trials began in 1995. In 1997, the Phase 2 clinical studies were initiated for men experiencing ED, then progressed to the Phase 3 trials that supported the drug’s FDA approval. Although Glaxo had an agreement with ICOS to share profits 50/50 for drugs resulting from the partnership, Glaxo let the agreement lapse in 1996 as the drugs developed were not in the company’s core markets.[3]
In 1998, ICOS Corporation and Eli Lilly and Company formed the Lilly ICOS, LLC, joint venture company to further develop and commercialize tadalafil as a treatment for ED. Two years later, Lilly ICOS, LLC, filed a new drug application with the FDA for compound IC351 (under the tadalafil generic name, and the Cialis brand name). In May 2002, Lilly ICOS reported to the American Urological Association that clinical trial testing demonstrated that tadalafil was effective for up to 36 hours, and one year later, the FDA approved tadalafil. One advantage Cialis has over Viagra and Levitra is its 17.5-hour half-life (thus Cialis is advertised to work for up to 36 hours, after which time there remains approximately 25 percent of the absorbed dose in the body) when compared to the four-hour half–life of sildenafil (Viagra).
In 2007, Eli Lilly and Company bought the ICOS Corporation for $2.3 billion. As a result, Eli Lilly owned Cialis and then closed the ICOS operations, ending the joint venture and firing most of ICOS’s approximately 500 employees, except for 127 employees of the ICOS biologics facility, which subsequently was bought by CMC Biopharmaceuticals A/S (CMC).

Tadalafil Molecule
Persons surnamed “Cialis” objected to Eli Lilly and Company’s so naming the drug, but the company has maintained that the drug’s trade name is unrelated to the surname.[4]
On October 6, 2011, the U.S. FDA approved tadalafil [5] to treat the signs and symptoms of benign prostatic hyperplasia (BPH). BPH is a condition in males in which the prostate gland becomes enlarged, obstructing the free flow of urine. Symptoms may include sudden urges to urinate (urgency), difficulty in starting urination (hesitancy), a weak urine stream, and more frequent urination- especially at night. The FDA has also approved tadalafil for treatment of both BPH and erectile dysfunction (ED) where the two conditions co-exist.
Although available since 2003 in 5, 10, 20 mg dosage, in late 2008/early 2009, the U.S. FDA approved the commercial sale of Cialis in 2.5 mg dosage as a one-a-day treatment for ED. The 2.5 mg dose avoids earlier dispensing restrictions on higher dosages. The price of the 5 mg and 2.5 mg are often similar, so some people score and split the pill.[6] The manufacturer does not recommend splitting.
Moreover, tadalafil (Adcirca) 40 mg was approved in 2009 in the United States and Europe (and 2010 in Canada and Japan) as a once-daily therapy to improve exercise ability in patients withpulmonary arterial hypertension. In patients with pulmonary arterial hypertension, the pulmonary vascular lumen is decreased as a result of vasoconstriction and vascular remodeling, resulting in increased pulmonary artery pressure and pulmonary vascular resistance. Tadalafil is believed to increase pulmonary artery vasodilation, and inhibit vascular remodeling, thus lowering pulmonary arterial pressure and pulmonary vascular resistance. Right heart failure is the principal consequence of pulmonary arterial hypertension.
On October 6, 2011, the U.S. FDA approved tadalafil [6] to treat the signs and symptoms of benign prostatic hyperplasia (BPH). BPH is a condition in males in which the prostate gland becomes enlarged, obstructing the free flow of urine. Symptoms may include sudden urges to urinate (urgency), difficulty in starting urination (hesitancy), a weak urine stream, and more frequent urination- especially at night. The FDA has also approved tadalafil for treatment of both BPH and erectile dysfunction (ED) where the two conditions co-exist.
Tadalafil has been used in approximately 15,000 men participating in clinical trials, and over eight million men worldwide (primarily in the post-approval/post-marketing setting). The most commonside effects when using tadalafil are headache, stomach discomfort or pain, indigestion, burping, acid reflux. back pain, muscle aches, flushing, and stuffy or runny nose. These side effects reflect the ability of PDE5 inhibition to cause vasodilation (cause blood vessels to widen), and usually go away after a few hours. Back pain and muscle aches can occur 12 to 24 hours after taking the drug, and the symptom usually disappears after 48 hours.
In May 2005, the U.S. Food and Drug Administration found that tadalafil (along with other PDE5 inhibitors) was associated with vision impairment related to NAION (nonarteritic anterior ischemic optic neuropathy) in certain patients taking these drugs in the post-marketing (outside of clinical trials) setting. Most, but not all, of these patients had underlying anatomic or vascular risk factors for development of NAION unrelated to PDE5 use, including: low cup to disc ratio (“crowded disc”), age over 50, diabetes, hypertension, coronary artery disease, hyperlipidemia and smoking. Given the small number of NAION events with PDE5 use (fewer than one in one million), the large number of users of PDE5 inhibitors (millions) and the fact that this event occurs in a similar population to those who do not take these medicines, the FDA concluded that they were not able to draw a cause and effect relationship, given these patients underlying vascular risk factors or anatomical defects. However, the label of all three PDE5 inhibitors was changed to alert clinicians to a possible association.
In May 2005, the U.S. Food and Drug Administration found that tadalafil (along with other PDE5 inhibitors) was associated with vision impairment related to NAION (nonarteritic anterior ischemic optic neuropathy) in certain patients taking these drugs in the post-marketing (outside of clinical trials) setting. Most, but not all, of these patients had underlying anatomic or vascular risk factors for development of NAION unrelated to PDE5 use, including: low cup to disc ratio (“crowded disc”), age over 50, diabetes, hypertension, coronary artery disease, hyperlipidemia and smoking. Given the small number of NAION events with PDE5 use (fewer than one in one million), the large number of users of PDE5 inhibitors (millions) and the fact that this event occurs in a similar population to those who do not take these medicines, the FDA concluded that they were not able to draw a cause and effect relationship, given these patients underlying vascular risk factors or anatomical defects. However, the label of all three PDE5 inhibitors was changed to alert clinicians to a possible association.
In October 2007, the FDA announced that the labeling for all PDE5 inhibitors, including tadalafil, requires a more prominent warning of the potential risk of sudden hearing loss as the result of postmarketing reports of deafness associated with use of PDE5 inhibitors.[7]
Selectivity compared with other PDE5 inhibitors
Tadalafil, sildenafil, and vardenafil all act by inhibiting the PDE5 enzyme. These drugs also inhibit other PDE enzymes. Sildenafil and vardenafil inhibit PDE6, an enzyme found in the eye, more than tadalafil.[9] Some sildenafil users see a bluish tinge and have a heightened sensitivity to light because of PDE6 inhibition.[3] Sildenafil and vardenafil also inhibit PDE1 more than tadalafil.[9]PDE1 is found in the brain, heart, and vascular smooth muscle.[9] It is thought that the inhibition of PDE1 by sildenafil and vardenafil leads to vasodilation, flushing, and tachycardia.[9] Tadalafil inhibits PDE11 more than sildenafil or vardenafil.[9] PDE11 is expressed in skeletal muscle, the prostate, the liver, the kidney, the pituitary gland, and the testes.[9] The effects on the body of inhibiting PDE11 are not known.[9]

20 mg Cialis tablet
In the United States, the FDA relaxed rules on prescription drug marketing in 1997, allowing advertisements targeted directly to consumers.[10] Lilly-ICOS hired the Grey Worldwide Agency in New York, part of the Grey Global Group, to run the Cialis advertising campaign.[11] Marketers for Cialis has taken advantage of its greater duration compared to its competitors in advertisements for the drug; Stuart Elliot of The New York Times opined: “The continuous presence of women in Cialis ads is a subtle signal that the drug makes it easier for them to set the pace with their men, in contrast to the primarily male-driven imagery for Levitra and Viagra.”[11] Iconic themes in Cialis ads include couples in bathtubs and the slogan “When the moment is right, will you be ready?”[11] Cialis ads were unique among the ED drugs in mentioning specifics of the drug.[12] As a result, Cialis ads were also the first to describe the side effects in an advertisement, as the FDA requires advertisements with specifics to mention side effects. One of the first Cialis ads aired at the 2004 Super Bowl.[12] Just weeks before the Super Bowl, the FDA required more possible side effects to be listed in the advertisement, including priapism.[12] Although many parents objected to the Cialis ad being aired during the Super Bowl, Janet Jackson‘s halftime “wardrobe malfunction” overshadowed Cialis.[12] In January 2006, the Cialis ads were tweaked, adding a doctor on screen to describe side effects and only running ads where more than 90 percent of the audience are adults, effectively ending Super Bowl ads.[10] In 2004, Lilly-ICOS, Pfizer, and GlaxoSmithKline spent a combined $373.1 million to advertise Cialis, Viagra, and Levitra respectively.[12] Cialis has sponsored many golf events, including the America’s Cup and the PGA Tour, once being title sponsor of the PGA Tour Western Open tournament.[13]
CIALIS (tadalafil) is a selective inhibitor of cyclic guanosine monophosphate (cGMP)-specific phosphodiesterase type 5 (PDE5). Tadalafil has the empirical formula C22H19N3O4 representing a molecular weight of 389.41. The structural formula is:
 |
The chemical designation is pyrazino[1',2':1,6]pyrido[3,4-b]indole-1,4-dione, 6-(1,3-benzodioxol-5-yl)2,3,6,7,12,12a-hexahydro-2-methyl-, (6R,12aR)-. It is a crystalline solid that is practically insoluble in water and very slightly soluble in ethanol.
CIALIS is available as almond-shaped tablets for oral administration. Each tablet contains 2.5, 5, 10, or 20 mg of tadalafil and the following inactive ingredients: croscarmellose sodium, hydroxypropyl cellulose, hypromellose, iron oxide, lactose monohydrate, magnesium stearate, microcrystalline cellulose, sodium lauryl sulfate, talc, titanium dioxide, and triacetin.
Tadalafil, (6R-trans)-6-(l,3-benzodioxol-5-yl)-2,3,6,7,12,12a-hexahydro-2- methyl-pyrazino[r,2':l,6]pyrido[3,4-b]indole-l,4-dione, with the structural formula shown below, is a white crystalline powder. (CAS# 171596-29-5). Tadalafil is a potent and selective inhibitor of the cyclic guanosine monophosphate (cGMP) – specific phosphodiesterase enzyme, PDE5. The inhibition of PDE5 increases the amount of cGMP, resulting in smooth muscle relaxation and increased blood flow. Tadalafil is therefore currently used in the treatment of male erectile dysfunction, and is commercially available as CIALIS ®.

Tadalafil U.S. Patent No. 5,859,006 describes the synthesis of tadalafil via the cyclization of TDCL (i.e., cis-methyl l,2,3,4-tetrahydro-2-chloroacetyl-l-(3,4- methylenedioxyphenyl)-9H-pyrido[3,4-b]mdole-3-carboxylate) using methylamine by purification by flash chromatography, followed by subsequent crystallization from methanol. Crude tadalafil typically requires additional purification steps, such as multiple extractions, crystallization, and/or flash chromatography, to remove the impurities present in the compound after synthesis is complete. Such purification processes increase the cost of producing tadalafil. Also, when repeating the US ’006 process, about 250 volumes of methanol were necessary for the crystallization step
Tadalafil can be prepared via a series of intermediates. One synthesis for preparing tadalafil is illustrated below in Scheme I:
SCHEME I
U.S. Patent No. 5,859,006 discloses the synthesis of a tadalafil intermediate
(Compound III) from D-tryptophan methyl ester (Compound II) and piperonal (Compound
I) using trifluoroacetic acid and dichloromethane, a halogenated solvent. Compound III is then reacted with chloroacetyl chloride (Compound IV) and chloroform to provide another intermediate of tadalafil (Compound V).
WO 2004/011463 discloses a process of preparing tadalafil intermediates from D-tryptophan methyl ester HCl salt and piperonal by refluxing the reagents in isopropyl alcohol, reacting the intermediate thus obtained with chloroacetyl chloride and tetrahydrofuran (THF) to provide another intermediate of tadalafil.
WO 2006/110893 discloses a process for the preparation of methyl ester intermediate (Compound III), and tadalafil using the methyl ester intermediate (CompoundII).
U.S. Patent Application Publication No. 2006/0258865 Al discloses a synthesis of the tadalafil intermediate (Compound III) from D-tryptophan methyl ester
(Compound II) and piperonal (Compound I) using a dehydrating agent selected from Na2SO4, K2SO4, MgSO4, CaSO4, CaCl2, molecular sieve or mixtures thereof and a high boiling solvent such as N,N-Dimethyl acetamide. Compound III is then reacted with chloroacetyl chloride (Compound IV) in the presence of a base such as NaHCO3 and an organic solvent such as dichloromethane, providing another intermediate of tadalafil (Compound V), which is further reacted with aqueous methyl amine solution to provide tadalafil.
………………………………………….
Scheme II and III.
……………………………………………………………………………………………………a compound of .Formula I
SCHEME III
SCHEME IV
Compound – 1 Compound – II
EXAMPLE l
The reaction scheme of this example is generally shown below in SchemeIV.
SCHEME IV
Compound – 1 Compound – II
Into a clean dry glass flask charged with ethanol (250 ml) under a nitrogen atmosphere was added Compound 1 (25 g) under stirring. The reaction mass was cooled to 0 to 50C and monomethylamine gas was purged into the reaction mixture for about 2 hours while maintaining the temperature between 0 to 50C. The temperature was raised to 75 to  0C and the reaction mixture was stirred under reflux for 2 hours. The reaction mixture was then cooled to 0 to 5°C and monomethylamine gas was again purged into the reaction mixture at 0 to 5°C. The temperature was again raised to 75 to 800C and stirred for about 1 hour. The reaction mixture was concentrated under vacuum to about 1/3 its original volume, cooled to 5 to 1O0C and stirred for 1 hour at this temperature. The solids were filtered and washed with chilled ethanol (50 ml). The wet solids were dried under vacuum for 6 hours.
0C and the reaction mixture was stirred under reflux for 2 hours. The reaction mixture was then cooled to 0 to 5°C and monomethylamine gas was again purged into the reaction mixture at 0 to 5°C. The temperature was again raised to 75 to 800C and stirred for about 1 hour. The reaction mixture was concentrated under vacuum to about 1/3 its original volume, cooled to 5 to 1O0C and stirred for 1 hour at this temperature. The solids were filtered and washed with chilled ethanol (50 ml). The wet solids were dried under vacuum for 6 hours.
0C and the reaction mixture was stirred under reflux for 2 hours. The reaction mixture was then cooled to 0 to 5°C and monomethylamine gas was again purged into the reaction mixture at 0 to 5°C. The temperature was again raised to 75 to 800C and stirred for about 1 hour. The reaction mixture was concentrated under vacuum to about 1/3 its original volume, cooled to 5 to 1O0C and stirred for 1 hour at this temperature. The solids were filtered and washed with chilled ethanol (50 ml). The wet solids were dried under vacuum for 6 hours.
Yield: 25g; Mp: 202-206.70C
Specific rotation (25°C) :+44.0 ( C=l% in DMSO)
13C NMR, DMSO-D6 : 25.78, 25.92, 57.89, 57.98, 101.17, 108.09, 108.32,
109.08, 111.48, 117.82, 118.62, 122.23, 122.97, 126.97, 135.97, 136.22, 136.55, 146.99,
147.48, 173.13
1H NMR, DMSO-D6, 300 MHz, Delta values: 2.6(m,lH), 2.7(m,3H),
2.8(d,lH), 3.0(d,lH), 3.6(bs,lH), 5.1(m,lH), 6.0(s,3H), 6.9-7.1(m, 5H), 7.2(d,lH),
7.4(d,lH), 7.8(bs, IH), 10.3(s, IH)
EXAMPLE 2
The reaction scheme of this example is generally shown below in SchemeV.
SCHEME V
Formula III Formula II
Into a clean dry flask charged with dichloromethane (200 ml) was added
Compound II (25 g) obtained in Example 1 under stirring at 25 to 300C. Next, triethylamine (16.11 g) was added to the reaction mixture and stirred for 30 minutes at 20 to 300C. The reaction mixture was cooled to 0 to 5°C and a solution of chloroacetyl chloride (12.93 g) in chloroform (50 ml) was added to the reaction mixture while maintaining temperature between -5 to 50C. The reaction mixture was stirred at -5 to 5°C for about 2 hours. Saturated aqueous sodium bicarbonate solution (50 ml) was added to the reaction mass slowly and the temperature of the reaction mixture was raised to 25 to 300C. The lower organic layer was separated and washed twice with water (75 ml). The chloroform extract was dried over anhydrous sodium sulfate. The organic layer was concentrated under vacuum until a thick yellow slurry was obtained. The slurry was cooled to 0 to5°C. The solids obtained were filtered and washed with 50 ml chilled chloroform. The wet product was dried at 750C under vacuum for 6 hours.
Yield: 22.5 g; HPLC Purity: 97%; Mp: 180-1820C
Specific rotation(25°C): -154.3(C=1% in DMSO)
13C. NMR(DMSO-Do, 300 MHZ)= 21.11, 25.88, 44.207, 51.60, 53.95,
101.16,107.66 109.56, 111.38, 118.36, 118.75, 121.58,122.74, 126.30, 130.31, 134.13,
136.57, 146.66, 147.03,167.43, 168.45
1H. NMR (CDC13, 300 MHZ):2.4(bs,3H), 3.1(m,lH), 3.8(m,lH),
4.3(bs,2H), 4.9(m,lH), 5.4(m,lH), 5.9(s,2H), 6.6-6.8(m,3H), 6.9(bs,lH), 7.1-7.3(m,3H),
7.6(d, IH), 7.7(bs,lH)
1H. NMR (DMSO-D6, 300 MHZ): 2.0 (bs,3H), 2.9(m,lH), 3.4(m,lH),
4.5(m,lH), 4.8(m,lH), 4.9(m,lH), 6.0(m,2H), 6.4-6.8(m,4H), 6.9-7.2(m,2H), 7.3(d, IH),
7.4(bs,lH), 7.5(d,lH), 10.8(s,lH)
EXAMPLE 3
The reaction scheme of this example is generally shown below in SchemeVI.
SCHEME VI
Formula II Formula I
Into a clean dry round bottom (RB) flask was charged tetrahydrofuran
(THF) (175 ml) under a nitrogen blanket and then cooled to -35 to -400C. Next 92 ml n- butyllithium (1.6 m solution in hexane) was added while maintaining the temperature between -35 to -400C. After the addition was complete, the reaction mixture was stirred at -35 to -400C for 15 minutes. A solution of compound of formula II (22.5 g) obtained in Example 2 in THF (75 ml) was prepared and slowly added to the reaction mixture while maintaining the temperature between -35 to -400C. After the addition was complete, the reaction mixture was stirred at -35 to -400C for 2.5 hours. Saturated aqueous ammonium chloride solution (25 ml) and 50 ml ethyl acetate was added to the reaction mixture at -35 to -400C. The temperature was raised to 25 to 300C and the two layers formed were separated. The upper organic layer was collected. The lower aqueous layer was thrice extracted with ethyl acetate (25 ml). The organic layers were combined together and washed with water (50 ml). The organic extract was dried over anhydrous sodium sulfate and concentrated under vacuum to obtain crude tadalafil as a dark brown solid. [0058] Yield: 22 g; HPLC Purity: 50%.
EXAMPLE 4
Purification of crude tadalafil
The crude tadalafil (22 g) obtained in Example 3 was suspended in 110 ml methanol and stirred for 1 hour at 25 to 300C. The solids obtained were filtered and washed with 25 ml chilled methanol. The wet product was dried at 600C under vacuum for 6 hours. This was further purified by using isopropyl alcohol. Yield: 9 g; HPLC Purity: >99.5%.
EXAMPLE 5
The reaction scheme of this example is generally shown below in SchemeVII.
Scheme VII
Formula VI where R = -OCH3 Formula VIA [0062] Into a clean dry RB flask charged with methanol (1900 ml) was added D- tryptophan methyl ester (190 g) under stirring at 25 to 300C. The reaction mixture was cooled to 0 to 50C. Monomethylamine gas was purged into the reaction mixture at 0 to 5°C for about 5-7 hours under stirring. The temperature of the reaction mixture was slowly raised to about 25 to 3O0C and stirred at this temperature for 5-7 hours. The reaction mixture was concentrated under vacuum to distill out the solvent. Diisopropyl ether (950 ml) was added and cooled to 25 to 3O0C under stirring for 1-2 hrs. The solids obtained were filtered, washed with Diisopropyl ether and dried under vacuum. [0063] MP: 122.4-1240C; Yield: 150 g (78.9 % w/w).
Specific rotation(25°C): +12.5 (C=I % in DMSO)
13 C NMR (300 MHZ,DMSO-D6): 25.71, 31.40, 55.67, 110.93, 111.55,
118.42, 118.73, 121.09, 123.95, 127.66,136.44, 175.39.
1H NMR (300 MHZ,DMSO-D6): 1.6(bs,2H), 2.5(m,3H), 2.8(m,lH),
3.1(m,lH), 3.4(m, IH), 6.9-7.2(m,3H), 7.3(d,lH), 7.5(d,lH), 7.8(bs,lH), 10.8(bs,lH)
EXAMPLE 6
The reaction scheme of this example is generally shown below in Scheme VIII.
SCHEME VIII
Formula VIA Formula VII
Into a clean, dry flask charged with methylene dichloride (MDC) (1000 ml) was added D-tryptophan methyl amide, the compound of Formula VIA (50 g), and piperonal, the compound of Formula VII (31.09 g), under stirring at 25 to 300C. The reaction mixture was cooled to 0 to 5°Cunder nitrogen atmosphere. Trifluoroacetic acid (85.3 g) was dissolved in MDC (250 ml) and the solution was slowly added to the reaction mixture at 0 to 5°C. The temperature of the reaction mixture was raised to 20 to 300C and stirred at this temperature for 14-16 hours. The reaction was monitored by TLC, workup was done as follows, the pH of the reaction mixture was adjusted to 8-9 using sodium carbonate solution under stirring, the two layers were settled, separated and the lower MDC layer was washed with water. The MDC layer was then dried over anhydrous sodium sulfate. The reaction mass was concentrated under vacuum at 40 to 5O0C to remove the solvent. The compound was precipitated using ethyl acetate, the solids were filtered, washed with ethyl acetate and dried.
Yield: 52.5 g; Yield: 105% w/w, HPLC Purity: 71% cis and 27% trans isomer (HPLC).
EXAMPLE 7
The reaction scheme of this example is generally shown below in Scheme IX.
SCHEME IX
1]CICOCH2C1 2]crystn
Formula m
Formula H
Into a clean dry flask charge with dichloromethane (400 ml) under a nitrogen atmosphere was added the compound of Formula III obtained in Example 6 and triethylamine (28.96 g) under stirring at 20 to 3O0C. The reaction mixture was then cooled to 0 to 50C. A mixture of chloroacetyl chloride (25.85 g) in dichloromethane (100 ml) was prepared and slowly added to the reaction mixture while maintaining the temperature between -5 to 50C in 1-2 hrs. The reaction mixture was stirred at 0 to 50C for 30 min and then saturated sodium bicarbonate solution (100 ml) was added at 5 to 100C under stirring. The temperature of the reaction mixture was raised to 25 to 300C and stirred at this temperature for 15 minutes. The layers were then separated. The lower MDC layer was collected, washed twice with 100 ml water and dried over anhydrous sodium sulfate. The
MDC layer was concentrated to distill out MDC until a stirrable mass was left behind. The mass was cooled to 25-3O0C and filtered, washed, to yield off-white to light yellow colored solids. The resulted product was the cis isomer, the trans isomer left behind in the mother liquor.
Yield = 25.5 g (50%w/w); HPLC Purity: > 97%.
The physical and spectral data was similar to that obtained in Example 2.
EXAMPLE 8
The reaction scheme of this example is generally shown below in SchemeX.
SCHEME X
Formula II
Into a clean dry round bottom (RB) flask was charged THF (1625 ml) under a nitrogen blanket and then cooled to -35 to -400C. Next, 505 ml n-butyllithium (1.6 m solution in hexane) was added while maintaining the temperature between -35 to -4O0C. After the addition was complete, the reaction mixture was stirred at -35 to -4O0C for 15 minutes. 72 ml diisopropyl amine was then added at -35 to -400C and then stirred at 0-50C for 1 hr. A solution of Compound of formula II (125 g) obtained in Example 7 in THF (625 ml) was prepared and slowly added to the reaction mixture while maintaining the temperature between -40 to -5O0C. After the addition was complete, the reaction mixture was stirred at -35 to -400C for 2-6 hours. Saturated aqueous ammonium chloride solution (250 ml) and ethyl acetate (125 ml) was added to the reaction mixture at -35 to -400C. The temperature was raised to 25 to 300C and the two layers formed were separated. The upper organic layer was collected. The lower aqueous layer was extracted with ethyl acetate (65 ml). The organic layers were combined together and distilled. Isopropyl alcohol (1250 ml) was added and the distillation was continued. A mixture of methanol (250 ml) and isopropanol (375 ml) were added and crude tadalafϊl was obtained upon cooling. The crude product was filtered, washed with water and dried. [0076] Yield: 60 g; (48% w/w); HPLC Purity: >99%.
EXAMPLE 9
Purification of crude Tadalafil
The crude tadalafil obtained in Example 8 was suspended in methanol (600 ml) and stirred for 1 hour at reflux. The mixture was cooled and the solids obtained were filtered and washed with chilled methanol (60 ml). The wet product was dried at under vacuum.
Yield: 56 g; HPLC Purity: 99.8%.
……………………………………………………………………………………
Beilstein J. Org. Chem. 2011, 7, 442–495.
A different approach was used in the synthesis of the phosphodiesterase inhibitor tadalafil (132, Cialis) starting from commercially available (D)-tryptophan methyl ester to form the indolopiperidine motif 135 via a Pictet–Spengler reaction followed by a double condensation to install the additional diketopiperazine ring (Scheme 28) [38,39].
![[1860-5397-7-57-i28]](http://www.beilstein-journals.org/bjoc/content/inline/1860-5397-7-57-i28.png?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 28: Optimised Pictet–Spengler reaction towards tadalafil.
To achieve the high levels of cis selectivity required from the Pictet–Spengler reaction, an extensive investigation of solvents and the influence of additives was undertaken [40]. It was identified that the use of a specific 23 mol % of benzoic acid significantly increased the cis/trans ratio from a base level of 55:45 to 92:8 (16 h reaction time at ambient temperature) in an overall yield of 86%. It was also determined that more polar solvents such as acetonitrile and nitromethane preferentially solvated the trans product and thereby allowed the isolation of the ciscompound by precipitation. It was also shown that by heating the reaction mixture under reflux the product distribution could be driven to the thermodynamically more favoured cis isomer having both the ester and the piperonyl moiety in equatorial positions. Hence, after heating under reflux for 8 h the cis/trans ratio was found to be 99:1 and the product could be isolated in an overall yield of 91%. This work represents an impressive example of a well considered and executed process optimisation study.
………………………………
The process disclosed in the patent US 5 859 006 (Scheme 1) involves condensation of D-tryptophan methyl ester with a piperonal derivative to yield a compound of formula (II). After conversion into a thioamide derivative of formula (III), cyclization occurs in presence of both an alkylating and reducing agents to provide a tetrahydro-β-carboline derivative of formula (IV), which on treatment with chloroacetyl chloride and methyl amine, gives Tadalafil. The compound of formula (IV) can also be obtained in one step, after separation of the other diastereoisomer, by a Pictet Spengler reaction between D-tryptophan methyl ester and piperonal in presence of an acid, such as trifluoroacetic acid.
- The patent application WO2007/10038 discloses the reaction of D-tryptophan with piperonal to provide a tetrahydro-β-carboline acid that was cyclised to Tadalafil in presence of a sarcosine derivative.
The patent application WO2007/1107 discloses the reaction of D-tryptophan methyl amide with piperonal, to provide an intermediate that after reaction with chloroacetyl chloride cyclises to Tadalafil in presence of butyllithium.
Thus, the active substance prepared by the processes known up till now can only be obtained in a satisfactory quality after running through a large number of process steps. Moreover a toxic alkylating agent, such as methylamine, is often used.

Example 1
- (1R,3R)-methyl-1,2,3,4-tetrahydro-2-(2-(benzyl(methyl)amino)acetyl)-1-(3,4-methylenedioxyphenyl)-9H-pyrido[3,4-b]indole-3-carboxylate (VII)
- A 50 mL three-necked flask fitted with thermometer and reflux condenser was charged with (1R,3R)-methyl 1,2,3,4-tetrahydro-2-chloroacetyl-1-(3,4-methylenedioxyphenyl)-9H-pyrido [3,4-b] indole-3-carboxylate (VI) (1.39 g, 3.26 mmol), DMA (5.33 mL), K2CO3 (0.5 g, 3.6 mmol) and N-benzylmethylamine (0.41 mL, 3.26 mmol). The resultant solution was stirred at room temperature. After 2 hours, the mixture was poured in brine (20 mL) and extracted with isopropyl acetate. The combined organic phases were washed with brine (3 x 5 mL), dried over sodium sulfate and concentrated to a residue under reduced pressure, affording 1.5 g of the desired product (VII), as a white solid. Yield: 70%.
1H NMR (d6-DMSO 300 MHz, 298K) 2.24 (s, 3H), 2.94-3.00 (m, 5H), 3.44-3.68 (m, 3H), 5.56 (bd, J = 6.4, 1H), 5.95 (s, 1H), 5.96 (s, 1H), 6.55 (bd, J = 7.4, 1H), 6.75 (bs, 1H), 6.77 (d, J = 8.0, 1H), 6.84 (bs, 1H), 7.05 (td, J = 7.4, 0.9, 1H), 7.12 (td, J = 7.5, 1.2, 1H), 7.17-7.32 (m, 6H), 7.56 (d, J = 7.7, 1H), 10.76 (bs, 1H)
13C NMR (d6-DMSO 75.4 MHz, 298K) 21.9, 42.5, 51.3, 51.9, 52.4, 61.0, 61.7, 101.5, 107.0, 108.0, 109.8, 111.8, 118.5, 119.2, 122.0, 123.0, 126.7, 127.7, 128.7, 129.6, 131.1, 134.7, 137.1, 138.6, 147.1, 147.5, 170.6, 171.5

Example 2
- (6R-trans)-6-(1,3-benzodioxol-5-yl)-2,3,6,7,12,12a-hexahydro-2-methyl-pyrazino [1',2':1,6] pyrido [3,4-b] indole-1,4-dione (Tadalafil) (I)Under H2 atmosphere (3 atm) and magnetic stirring, Raney® Ni (2800 slurry in water, 0.0276 g, 0.47 mmol), previously washed with methanol (3 times), was added to a solution of (1R,3R)-methyl-1,2,3,4-tetrahydro-2-(2-(benzyl(methyl)amino)acetyl)-1-(3,4-methylenedioxyphenyl)-9H-pyrido[3,4-b]indole-3-carboxylate (VII) (3.00 g, 4.70 mmol) in DMA (21.3 mL). The mixture was heated at 90°C for 17 hours and then cooled to room temperature. The suspension was filtered over a pad of Celite® and the resulting solutionand the resulting solution was concentrated until 6 mL. Methanol (12 mL) was added and the solid which was so obtained was filtered over Buchner, washed with methanol (4 mL) and oven-dried under reduced pressure for 2 hours, affording 1.3 g of the title compound, as a white solid. Yield: 70%
1H NMR (d6-DMSO 300 MHz, 298K): 2.91-3.00 (m, 4H), 3.32 (s, 1H), 3.47-3.54 (dd, J = 4.6, 11.3, 1H), 3.93 (d, J = 17.1, 1H), 4.17 (d, J = 17.1, 1H), 4.35-4.40 (dd, J = 4.27, 11.6, 1H), 5.91 (s, 2H), 6.11 (s, 1H), 6.76 (s, 2H), 6.85 (s, 1H), 6.98-7.06 (m, 2H), 7.28 (d, J = 7.9, 1H), 7.52 (d, J = 7.3, 1H), 11.0 (s, 1H)
13C NMR (d6-DMSO 75.4 MHz, 298K) 23.8, 33.4, 52.0, 55.9, 56.1, 101.5, 105.3, 107.6, 108.6, 111.9, 118.7, 119.5, 119.9, 121.8, 126.4, 134.5, 136.8, 137.6, 146.7, 147.6, 167.1, 167.5

References
- Daugan, A; Grondin P, Ruault C, Le Monnier de Gouville AC, Coste H, Kirilovsky J, Hyafil F, Labaudinière R (October 9, 2003). “The discovery of tadalafil: a novel and highly selective PDE5 inhibitor. 1: 5,6,11,11a-tetrahydro-1H-imidazo[1',5':1,6]pyrido[3,4-b]indole-1,3(2H)-dione analogues”. Journal of Medicinal Chemistry 46 (21): 4525–32. doi:10.1021/jm030056e . PMID 14521414.
- Richards, Rhonda (September 17, 1991). “ICOS At A Crest On Roller Coaster”. USA Today. p. 3B.
- Ervin, Keith (June 21, 1998). “Deep Pockets + Intense Research + Total Control = The Formula — Bothell Biotech Icos Keeps The Pipeline Full Of Promise”. The Seattle Times. p. F1. Retrieved January 10, 2009.
- Revill, Jo (February 2, 2003). “Drugs giant says its new pill will pack more punch than rival Viagra”. The Observer. Retrieved 2007-04-06.
- http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm274642.htm
- https://www.consumerreports.org/health/resources/pdf/best-buy-drugs/money-saving-guides/english/PillSplitting-FINAL.pdf
- “FDA Announces Revisions to Labels for Cialis, Levitra and Viagra”. Food and Drug Administration. 2007-10-18. Retrieved 2009-09-28.
- “Cialis: Warnings, Precautions, Pregnancy, Nursing, Abuse”. RxList. 2007. Retrieved 2007-04-06.
- Bischoff, E (June 2004). “Potency, selectivity, and consequences of nonselectivity of PDE inhibition”. International Journal of Impotence Research 16: S11–4.doi:10.1038/sj.ijir.3901208 . PMID 15224129. Retrieved January 19, 2009.
- Elliott, Stuart (January 10, 2006). “For Impotence Drugs, Less Wink-Wink”. The New York Times. p. C2. Retrieved January 15, 2009.
- Elliott, Stuart (April 25, 2004). “Viagra and the Battle of the Awkward Ads”. The New York Times. p. 1. Retrieved January 15, 2009.
- McCarthy, Shawn (March 5, 2005). “First they tried to play it safe; Ads for erectile dysfunction drug Cialis bared all – including a scary potential side effect. It was risky but it has paid off”. The Globe and Mail. p. B4.
- Loyd, Linda (July 6, 2003). “Two Pills Look to Topple Viagra’s Reign in Market; Levitra Expects Approval Next Month, Cialis Later This Year”. The Philadelphia Inquirer. p. E01.
- 38 is 1 below
- 39 is 2 below
- 40 is 3 below
- daugan, A. C.-M. Tetracyclic Derivatives; Process of Preparation and Use. U.S. Patent 5,859,006, Jan 12, 1999.
- Daugan, A. C.-M. Tetracyclic Derivatives, Process of Preparation and Use. U.S. Patent 6,025,494, Feb 15, 2000.
- Shi, X.-X.; Liu, S.-L.; Xu, W.; Xu, Y.-L. Tetrahedron: Asymmetry 2008, 19, 435–442.doi:10.1016/j.tetasy.2007.12.017
DAUGAN A ET AL: “THE DISCOVERY OF TADALAFIL: A NOVEL AND HIGHLY SELECTIVE PDE5 INHIBITOR. 2: 2,3,6,7,12,12A-HEXAHYDROPYRAZINO[1′,2′:1,6 ÜPYRIDO[3,4-B ÜINDOLE-1,4-DIONE” JOURNAL OF MEDICINAL CHEMISTRY, AMERICAN CHEMICAL SOCIETY. WASHINGTON, US, vol. 46, no. 21, 2003, pages 4533-4542, XP008052656 ISSN: 0022-2623
- Tadalafil bound to proteins in the PDB
- National Institutes of Health – Medlineplus
- Material Safety Data Sheet PDF file
- Official Cialis (Tadalafil) Website
- U.S. National Library of Medicine: Drug Information Portal – Tadalafil
| WO2009004557A2 * | Jun 28, 2008 | Jan 8, 2009 | Ranbaxy Lab Ltd | A process for the preparation of intermediates of tetracyclic compounds |
| WO2009148341A1 | Jun 3, 2009 | Dec 10, 2009 | Zaklady Farmaceutyczne Polpharma Sa | Process for preparation of tadalafil |
| WO2012107549A1 | Feb 10, 2012 | Aug 16, 2012 | Interquim, S.A. | PROCESS FOR OBTAINING COMPOUNDS DERIVED FROM TETRAHYDRO-ß-CARBOLINE |
| EP2107059A1 | Mar 31, 2008 | Oct 7, 2009 | LEK Pharmaceuticals D.D. | Conversion of tryptophan into ß-carboline derivatives |
| US8445698 | Jun 28, 2008 | May 21, 2013 | Ranbaxy Laboratories Limited | Process for the preparation of an intermediate of tadalafil |
..................................
This is a very interesting topic for me. thanks for the links. I'll go and look. Look here https://writepaperfor.me/.
ReplyDeletegeneric medications at lowest price
ReplyDeleteThanks for this amazing and useful information. Hope you’ll like mine… Edcarerx.com is an online pharmacy store that sells the best Sildenafil Citrate – Generic Viagra – Generic ED medication to men suffering with erectile dysfunction, Impotency problem and premature ejaculation. We provide medicines at best reasonable price in USA. We have built up ourselves as a leader in the category of online pharmacies since the inception our operation. We have taken continuous efforts towards providing quality and high standard products to our customers by confirming to the standards of manufacturing and quality control. Before providing the medications we rectify the medicines through testing and checking of the products, the medications sold by us are approved by the FDA (Food and Drug Administration) due to the existence of the active ingredients in the product.
The long term affects of the cobra 120mg are still being investigated but no serious side effects of this drug have been reported so far.
ReplyDelete