CONAZOLE SERIES
PART 1
1 TERCONAZOLE
2 KETOKONAZOLE 2S 4R
3 ISAVUCONAZOLE
4 TIOCONAZOLE
5 FOSRAVUCONAZOLE
6 RAVUCONAZOLE
7 EFINACONAZOLE
8 ALBACONAZOLE
9 BUTOCONAZOLE
SEE PART 2 ............http://apisynthesisint.blogspot.in/p/blog-page.html
10
11
12

1 TERCONAZOLE












































PART 1
1 TERCONAZOLE
2 KETOKONAZOLE 2S 4R
3 ISAVUCONAZOLE
4 TIOCONAZOLE
5 FOSRAVUCONAZOLE
6 RAVUCONAZOLE
7 EFINACONAZOLE
8 ALBACONAZOLE
9 BUTOCONAZOLE
SEE PART 2 ............http://apisynthesisint.blogspot.in/p/blog-page.html
10
11
12
1 TERCONAZOLE
 | |
| SYSTEMATIC (IUPAC) NAME | |
|---|---|
| 1-[4-[ [(2S,4S)-2-(2,4-Dichlorophenyl)-2- (1,2,4-triazol-1-ylmethyl)- 1,3-dioxolan-4-yl]methoxy]phenyl]- 4-propan-2-yl-piperazine | |
| CLINICAL DATA | |
| TRADE NAMES | Terazol |
| AHFS/DRUGS.COM | monograph |
| MEDLINEPLUS | a688022 |
| LEGAL STATUS | ? |
| PHARMACOKINETIC DATA | |
| PROTEIN BINDING | 94.9% |
| IDENTIFIERS | |
| CAS NUMBER | 67915-31-5 |
| ATC CODE | G01AG02 |
| PUBCHEM | CID 441383 |
| DRUGBANK | DB00251 |
| CHEMSPIDER | 390122 |
| UNII | 0KJ2VE664U |
| KEGG | D00888 |
| CHEMBL | CHEMBL1306 |
| CHEMICAL DATA | |
| FORMULA | C26H31Cl2N5O3 |
| MOL. MASS | 532.462 g/mol |
Terconazole is an anti-fungal medication, primarily used to treat vaginal fungal infections.
The synthesis of racemic terconazole [J. Heeres et al., J. Med . Chem . , 26 , 611 11983)] is similar. differing in the introduction of a 1 H- 1 , 2,4-triazol-1-yl substituent in place of 1H-imidazol-1-yl and in the nature of the phenol used in the last step of the synthetic sequence, which phenol is 1-methylethyl-4-(4- hydroxyphenyl)piperazme instead of 1-acetyl-4-(4-nydroxyphenyl)piperazine.

Example 20: (2S,4R) -(-)-1-[4-[[2-(2,4-dichlorophenyl)-2-[(1H-1,2,4-triazol-1-yl]methyl-1,3-dioxolane-4-yl]methoxy]phenyl]-4-(1-methylethyl)piperazine, (2S,4R) – (-)-terconazole.
This compound is prepared following the process described for (+)-torconazole, starting from (2S,4S)-(-)-IV (Ar = 2,4-dichlorophenyl, Y = N, R = CH3) (224 mg, 0.55 mmol), 4-(4-hydroxyphenyl)-1-(1-methylethyl)-piperazine (121 mg, 0.55 mmol), NaH (22.4 mg, 0.56 mmol) in 8 ml of DMSO. (2S,4R) -(-(-terconazole ((2S,4R)-V, Ar
= 2,4-dichlorophenyl, Y = N, Z = CH(CH3)2) is obtained as a white solid, m.p. 76-78ºC, [α]D 20= -12.0 (c = 0.4.
CHCl3).
Example 17 : (2R,4S)-(+)-1-[4-[[2-(2,4-dichlorophenyl)- 2-[(1H-1,2,4-triazol-1-yl]methyl-1,3-dioxolane-4-yl]methyl]phenyl]-4-(1-methylethyl)piperazine, (2R,4S)-(+)-terconazole.
To a suspension of NaH (60-65% dispersion in paraffin, 36 mg, 0.90 mmol) in anhydrous DMSO (8 ml), 4-(4-hydroxyphenyl) -1 – ( 1-methyle thyl ) p iper az ine ( 193 mg , 0 . 88 mmol ) is added and the mixture is stirred for 1 hour at room temperature. Then, (2R,4R)-(+)-IV (Ar = 2,4-dichlorophenyl, Y = N, R = CH3 ) is added (180 mg, 0.44 mmol) and the mixture is heated at 80°C for 4 hours. The reaction mixture is allowed to cool to room temperature, diluted with water (20 ml) and extraoteo with CH2Cl2 (3 × 25 ml). The combined organic phases are washed with 5N NaOH (3 × 25 ml) and water (3 × 25 ml dried with Na2SO4 and the solvent is evaporated of: under vacuum. The oily residue thus obtained is crystallized from diisopropyl ether to give (2R,4S)-(+)-terconazole ((2R,4S)-V, Ar = 2,4-cichlorophenyl, Y = N, Z = CH(CH3)2) (140 mg, 59 % yield) as a white solid, m.p. 72-74’C, [α]D 20 = + 11,05 (c = 0.4, CHCl3).
IR (KBr), ʋ : 1585, 1512, 1454, 1380, 1270, 1239, 1137, 1048, 979, 820, 675 cm-1.
1H-NMR (200 MHz, CDCl3), δ : 1.11 [d, J=6.5 Hz, 5H, (CH3)2CH], 2.73 [m, 5H, 3-H2, 5-H2 and (CH3)2CH], 3.49
(dd, J=9.6 Hz, J’=6.3 Hz, 1H), 3.80 (m, 2H ) and 3.91
(dd, J=8.2 Hz, J’=6.6 Hz, 1H) (4′ ‘-CH2 and 5′ ‘-H2), 4.35
(m, 1H, 4′ ‘-H), 4.74 (d, J=14.6 Hz, 1H) and 4.84 (d, J=14.6 Hz, 1H) (CH2-N), 6.76 [d, J=9.0 Hz, 2H, C2'(6')- H], 6.88 [d, J=9.0 Hz, 2H, C3'(5')-H], 7.24 (dd, J=8.5
Hz, J’=2.0 Hz, 1H, 5”’-H), 7.46 (d, J=2.0 Hz, 1H,
3″‘-H), 7.56 (d, J=8.5 Hz, 1H, 6″‘-H), 7.89 (s, 1 H) and
8.20 (s, 1H) (triazole 3-H and 5-H).
Synthesis pathway
| SYNTHESIS A) |
|---|
     |
- DE 2804096 (Janssen; appl. 3.8.1978; prior. 31.1.1978).
- US 4,358,449 (Janssen; 9.11.1982; prior. 21.11.1977).
- US 4,144,346 (Janssen; 13.3.1979; prior. 21.11.1977, 31.1.1977).
- US 4,223,036 (Janssen; 16.9.1980; prior. 8.1.1979, 21.11.1977, 31.1.1977).
- Heeres, J. et al .: J. Med. Chem. (JMCMAR) 26, 611 (1983).
2 KETOKONAZOLE 2S 4R









KETOCONAZOLE 2S 4R
ALSO
142128-57-2
228850-16-6 (tartrate)
228850-16-6 (tartrate)
(-)-cis-1-Acetyl-4-[4-[2(S)-(2,4-dichlorophenyl)-2-(1H-imidazol-1-ylmethyl)-1,3-dioxolan-4(R)-ylmethoxy]phenyl]piperazine
531.431, C26 H28 Cl2 N4 O4
COR-003
DIO-902
LDKTZ
DIO-902
LDKTZ
CORTENDO
licensee DiObex
| Biological Role(s): | antifungal agent
An antimicrobial agent that destroys fungi by suppressing their ability to grow or reproduce. Antifungal agents differ from industrial fungicides in that they defend against fungi present in human or animal tissues.
|
| Application(s): | antifungal agent
An antimicrobial agent that destroys fungi by suppressing their ability to grow or reproduce. Antifungal agents differ from industrial fungicides in that they defend against fungi present in human or animal tissues.
|
Ketoconazole, 1-acetyl-4-[4-[[2-(2,4-dichlorophenyl)-2-[(1H-imidazol-1-yl)-methyl]-1,3– dioxolan-4-yl]methoxy]phenyl]piperazine, is a racemic mixture of the cis enantiomers (-)-(2S,4R) and (+)-(2R,4S) marketed as an anti-fungal agent. Ketoconazole inhibits fungal growth through the inhibition of ergosterol synthesis.(-)-Ketoconazole, the (2S,4R) enantiomer contained in the racemate of ketoconazole, is in phase III clinical trials at Cortendo for the treatment of endogenous Cushing’s syndrome. The company and licensee DiObex had also been developing the drug candidate for the treatment of type 2 diabetes; however, no recent development has been reported for this research.Preclinical studies have demonstrated the drug candidate’s ability to inhibit the synthesis of cortisol, resulting in substantial clinical benefits including lowering both blood pressure and cholesterol in addition to controlling glucose levels. It has also been shown that (-)-ketoconazole is responsible for virtually all of the cortisol synthesis inhibitory activity present in the racemate. Rights to the compound are shared with Cortendo.In 2012, orphan drug designation was assigned in the U.S. for the treatment of endogenous Cushing’s syndrome.
August 12, 2014 02:30 AM Eastern Daylight Time
GÖTEBORG, Sweden.–(BUSINESS WIRE)–Cortendo AB (OSE:CORT) today announced that the first patient has been enrolled into the Phase 3 SONICS trial, i.e., “Study Of NormoCort In Cushing’s Syndrome.”
“The enrollment of the first patient into the SONICS trial represents a significant milestone for Cortendo”
The patient was enrolled by one of the trial’s lead principal investigators at a Pituitary Center from a prestigious institution in Baltimore, Maryland. “The enrollment of the first patient into the SONICS trial represents a significant milestone for Cortendo”, said Dr. Theodore R Koziol. ”The SONICS clinical trial team is acutely focused on the implementation of the trial following a successful EU Investigator’s meeting in Barcelona in July, which we believe further solidified the foundation for the trial.”
Cortendo successfully completed its European Investigator meeting supporting SONICS held in Barcelona, Spain on July 17-18. More than 35 investigators/study coordinators, including many of the world’s leading Cushing’s experts from 24 study sites, were in attendance and received training for the trial. Based on the positive feedback from the meeting, Cortendo has gained further confidence that NormoCort (COR-003) has the potential to be an important future treatment option for patients afflicted with Cushing’s Syndrome. A second US Investigator meeting is also being planned for later this year.
”It was gratifying to participate in the NormoCort SONICS trial investigator meeting in my home town of Barcelona with so many esteemed colleagues dedicated to treating patients with Cushing’s Syndrome”, said Susan Webb M.D. Ph.D. Professor of Medicine Universitat Autonoma de Barcelona. ”There remains a significant unmet medical need for patients, and I am delighted to be part of the development of this new therapy”.
Cortendo has also further strengthened its internal as well as external teams to support the study and to position the trial for an increased recruitment rate. In July, Cortendo added both an experienced physician and internal Clinical Operations Director to the NormoCort development team. Cortendo, working in concert with its CROs supporting the SONICS trial, now has a team of approximately 20 personnel on the NormoCort development program.
Cortendo has previously communicated its plan to meet the recruitment goal by increasing the number of study sites from 38 to 45 worldwide. The company is at various levels of activation with more than 30 study sites to date. Therein, Cortendo expects a large proportion of the sites to be activated by the end of the third quarter this year and remains confident that essentially all sites will be open by the end of 2014.
Risk and uncertainty
The development of pharmaceuticals carries significant risk. Failure may occur at any stage during development and commercialization due to safety or clinical efficacy issues. Delays may occur due to requirements from regulatory authorities not anticipated by the company.
About Cortendo
Cortendo AB is a biopharmaceutical company headquartered in Göteborg, Sweden. Its stock is publicly traded on the NOTC-A-list (OTC) in Norway. Cortendo is a pioneer in the field of cortisol inhibition and has completed early clinical trials in patients with Type 2 diabetes. The lead drug candidate NormoCort, the 2S, 4R-enantiomer of ketoconazole, has been re-focused to Cushing’s Syndrome, and has entered Phase 3 development. The company’s strategy is to primarily focus its resources within orphan drugs and metabolic diseases and to seek opportunities where the path to commercialization or partnership is clear and relatively near-term. Cortendo’s business model is to commercialize orphan and specialist product opportunities in key markets, and to partner non-specialist product opportunities such as diabetes at relevant development stages.
Cortendo AB (publ)
Sweden: Box 47 SE-433 21 Partille Tel. / Fax: +46 (0)31-263010
USA: 555 East Lancaster Ave Suite 510 Radnor, PA 19087 Tel: +1 610-254-9200 Fax: +1 610-254-9245
This information was brought to you by Cision http://news.cision.com
Contacts
Alexander Lindström
Chief Financial Officer Office
+1 610 254 9200
Mobile : +1 917 349 7210
E-mail : alindstrom@cortendo.com
Chief Financial Officer Office
+1 610 254 9200
Mobile : +1 917 349 7210
E-mail : alindstrom@cortendo.com
- Ketoconazole, 1-acetyl-4- [4-[[2-(2,4-dichlorophenyl)-2-[(1H-imidazol-1-yl)-methyl]-1,3-dioxolan-4-yl] methoxy] phenyl] piperazine, is a racemic mixture of the cis enantiomers (-)-(2S, 4R) and (+)-(2R, 4S) marketed as an anti-fungal agent. Ketoconazole inhibits fungal growth through the inhibition of ergosterol synthesis. Ergosterol is a key component of fungal cell walls.
- More recently, ketoconazole was found to decrease plasma cortisol and to be useful, alone and in combination with other agents, in the treatment of a variety of diseases and conditions, including type 2 diabetes, Metabolic Syndrome (also known as the Insulin Resistance Syndrome, Dysmetabolic Syndrome or Syndrome X), and other medical conditions that are associated with elevated cortisol levels. SeeU.S. Patent Nos. 5,584,790 ; 6,166,017 ; and 6,642,236 , each of which is incorporated herein by reference. Cortisol is a stress-related hormone secreted from the cortex of the adrenal glands. ACTH (adenocorticotropic hormone) increases cortisol secretion. ACTH is secreted by the pituitary gland, a process activated by secretion of corticotropin releasing hormone (CRH) from the hypothalamus.
- Cortisol circulates in the bloodstream and activates specific intracellular receptors, such as the glucocorticoid receptor (GR). Disturbances in cortisol levels, synthetic rates or activity have been shown to be associated with numerous metabolic complications, including insulin resistance, obesity, diabetes and Metabolic Syndrome. Additionally, these metabolic abnormalities are associated with substantially increased risk of cardiovascular disease, a major cause of death in industrialized countries. See Mårin P et al., “Cortisol secretion in relation to body fat distribution in obese premenopausal women.” Metabolism 1992; 41:882-886, Bjorntorp, “Neuroendocrine perturbations as a cause of insulin resistance.” Diabetes Metab Res Rev 1999; 15(6): 427-41, and Rosmond, “Role of stress in the pathogenesis of the metabolic syndrome.” Psychoneuroendocrinology 2005; 30(1): 1-10, each of which is incorporated herein by reference.
- While ketoconazole is known to inhibit some of the enzymatic steps in cortisol synthesis, such as, for example, 17α hydroxylase (Wachall et al., “Imidazole substituted biphenyls: a new class of highly potent and in vivo active inhibitors of P450 17 as potential therapeutics for treatment of prostate cancer.” Bioorg Med Chem 1999; 7(9): 1913-24, incorporated herein by reference) and 11b-hydroxylase (Rotstein et al., “Stereoisomers of ketoconazole: preparation and biological activity.” J Med Chem 1992; 35(15): 2818-25) and 11β-hydroxy steroid dehydrogenase (11β-HSD) (Diederich et al., “In the search for specific inhibitors of human 11β-hydroxysteroid-dehydrogenases (11β-HSDs): chenodeoxycholic acid selectively inhibits 11β-HSD-L” Eur J Endocrinol 2000; 142(2): 200-7, incorporated herein by reference) the mechanisms by which ketoconazole decreases cortisol levels in the plasma have not been reported. For example, there is uncertainty regarding the effect of ketoconazole on the 11β-hydroxy steroid dehydrogenase (11β-HSD) enzymes. There are two 11β-HSD enzymes. One of these, 11β-HSD-I, is primarily a reductase that is highly expressed in the liver and can convert the inactive 11-keto glucocorticoid to the active glucocorticoid (cortisol in humans and corticosterone in rats). In contrast, the other, 11β-HSD-II, is primarily expressed in the kidney and acts primarily as an oxidase that converts active glucocorticoid (cortisol in humans and corticosterone in rats) to inactive 11-keto glucocorticoids. Thus, the plasma concentration of active glucocorticoid is influenced by the rate of synthesis, controlled in part by the activity of adrenal 11β-hydroxylase and by the rate of interconversion, controlled in part by the relative activities of the two 11β-HSD enzymes. Ketoconazole is known to inhibit these three enzymes (Diederich et al., supra) and the 2S,4R enantiomer is more active against the adrenal 11β-hydroxylase enzyme than is the 2R,4S enantiomer (Rotstein et al., supra). However, there are no reports describing the effect of the two ketoconazole enantiomers on either of 11β-HSD-I or 11β-HSD-II, so it is not possible to predict what effects, if any, the two different ketoconazole enantiomers will each have on plasma levels of the active glucocorticoid levels in a mammal.
- Ketoconazole has also been reported to lower cholesterol levels in humans (Sonino et al. (1991). “Ketoconazole treatment in Cushing’s syndrome: experience in 34 patients.” Clin Endocrinol (Oxf). 35(4): 347-52; Gylling et al. (1993). “Effects of ketoconazole on cholesterol precursors and low density lipoprotein kinetics in hypercholesterolemia.” J Lipid Res. 34(1): 59-67) each of which is incorporated herein by reference). The 2S,4R enantiomer is more active against the cholesterol synthetic enzyme 14 αlanosterol demethylase than is the other (2R,4S) enantiomer (Rotstein et al infra). However, because cholesterol level in a human patient is controlled by the rate of metabolism and excretion as well as by the rate of synthesis it is not possible to predict from this whether the 2S,4R enantiomer of ketoconazole will be more effective at lowering cholesterol levels.
- The use of ketoconazole as a therapeutic is complicated by the effect of ketoconazole on the P450 enzymes responsible for drug metabolism. Several of these P450 enzymes are inhibited by ketoconazole (Rotsteinet al., supra). This inhibition leads to an alteration in the clearance of ketoconazole itself (Brass et al., “Disposition of ketoconazole, an oral antifungal, in humans.” Antimicrob Agents Chemother 1982; 21(1): 151-8, incorporated herein by reference) and several other important drugs such as Glivec (Dutreix et al., “Pharmacokinetic interaction between ketoconazole and imatinib mesylate (Glivec) in healthy subjects.” Cancer Chemother Pharmacol 2004; 54(4): 290-4) and methylprednisolone (Glynn et al., “Effects of ketoconazole on methylprednisolone pharmacokinetics and cortisol secretion.” Clin Pharmacol Ther 1986; 39(6): 654-9). As a result, the exposure of a patient to ketoconazole increases with repeated dosing, despite no increase in the amount of drug administered to the patient. This exposure and increase in exposure can be measured and demonstrated using the “Area under the Curve” (AUC) or the product of the concentration of the drug found in the plasma and the time period over which the measurements are made. The AUC for ketoconazole following the first exposure is significantly less than the AUC for ketoconazole after repeated exposures. This increase in drug exposure means that it is difficult to provide an accurate and consistent dose of the drug to a patient. Further, the increase in drug exposure increases the likelihood of adverse side effects associated with ketoconazole use.
- [0008]Rotstein et al. (Rotstein et al., supra) have examined the effects of the two ketoconazole cis enantiomers on the principal P450 enzymes responsible for drug metabolism and reported “…almost no selectivity was observed for the ketoconazole isomers” and, referring to drug metabolizing P450 enzymes: “[t]he IC50 values for the cis enantiomers were similar to those previously reported for racemic ketoconazole”. This report indicated that both of the cis enantiomers could contribute significantly to the AUC problem observed with the ketoconazole racemate.
- One of the adverse side effects of ketoconazole administration exacerbated by this AUC problem is liver reactions. Asymptomatic liver reactions can be measured by an increase in the level of liver specific enzymes found in the serum and an increase in these enzymes has been noted in ketoconazole treated patients (Sohn, “Evaluation of ketoconazole.” Clin Pharm 1982; 1(3): 217-24, and Janssen and Symoens, “Hepatic reactions during ketoconazole treatment.” Am J Med 1983; 74(1B): 80-5, each of which is incorporated herein by reference). In addition 1:12,000 patients will have more severe liver failure (Smith and Henry, “Ketoconazole: an orally effective antifungal agent. Mechanism of action, pharmacology, clinical efficacy and adverse effects.” Pharmacotherapy 1984; 4(4): 199-204, incorporated herein by reference). As noted above, the amount of ketoconazole that a patient is exposed to increases with repeated dosing even though the amount of drug taken per day does not increase (the “AUC problem”). The AUC correlates with liver damage in rabbits (Ma et al., “Hepatotoxicity and toxicokinetics of ketoconazole in rabbits.” Acta Pharmacol Sin 2003; 24(8): 778-782 incorporated herein by reference) and increased exposure to the drug is believed to increase the frequency of liver damage reported in ketoconazole treated patients.
- Additionally, U.S. Patent No. 6,040,307 , incorporated herein by reference, reports that the 2S,4R enantiomer is efficacious in treating fungal infections. This same patent application also reports studies on isolated guinea pig hearts that show that the administration of racemic ketoconazole may be associated with an increased risk of cardiac arrhythmia, but provides no data in support of that assertion. However, as disclosed in that patent, arrhythmia had not been previously reported as a side effect of systemic racemic ketoconazole, although a particular subtype of arrhythmia, torsades de pointes, has been reported when racemic ketoconazole was administered concurrently with terfenadine. Furthermore several published reports (for example, Morganroth et al. (1997). “Lack of effect of azelastine and ketoconazole coadministration on electrocardiographic parameters in healthy volunteers.” J Clin Pharmacol. 37(11): 1065-72) have demonstrated that ketoconazole does not increase the QTc interval. This interval is used as a surrogate marker to determine whether drugs have the potential for inducing arrhythmia. US Patent Number 6,040,307 also makes reference to diminished hepatoxicity associated with the 2S,4R enantiomer but provides no data in support of that assertion. The method provided in US Patent Number 6,040,307 does not allow for the assessment of hepatoxicity as the method uses microsomes isolated from frozen tissue.
…………………………
- DIO-902 is the single enantiomer 2S,4R ketoconazole and is derived from racemic ketoconazole. It is formulated using cellulose, lactose, cornstarch, colloidal silicon dioxide and magnesium stearate as an immediate release 200 mg strength tablet. The chemical name is 2S,4R cis-1-acetyl-4-[4-[[2-(2,4-dichlorophenyl)-2-(1H-imidazol-1-ylmethyl)-1,3-dioxolan-4-yl] methoxyl]phenyl] piperazine, the formula is C26H28Cl2N4O4, and the molecular weight is 531.44. The CAS number is 65277-42-1, and the structural formula is provided below. The chiral centers are at the carbon atoms 2 and 4 as marked.
- [0132]Ketoconazole is an imidazole-containing fungistatic compound. DIO-902 is an immediate release tablet to be taken orally and formulated as shown in the table below.
Component Percentage 2S,4R ketoconazole;
DIO-90250% Silicified Microcrystalline Cellulose, NF
(Prosolv HD 90)16.5 Lactose Monohydrate, NF (316 Fast-Flo) 22.4 Corn Starch, NF (STA-Rx) 10 Colloidal Silicon Dioxide, NF (Cab-O-Sil M5P) 0.5 Magnesium Stearate, NF 0.6 The drug product may be stored at room temperature and is anticipated to be stable for at least 2 years at 25° C and 50% RH. The drug is packaged in blister packs.

ketoconazole 2S,4R enantiomer
ketoconazole 2S,4S enantiomer
- ketoconazole 2R,4R enantiomer
ketoconazole 2R,4S enantiomer
……………………..
Journal of Medicinal Chemistry (Impact Factor: 5.61). 08/1992; 35(15):2818-25. DOI: 10.1021/jm00093a015
…………………….
Enantioselective separation of ketoconazole enantiomers by membrane extraction
A new process has been developed to separate ketoconazole (KTZ) enantiomers by membrane extraction, with the oppositely preferential recognition of hydrophobic and hydrophilic chiral selectors in organic and aqueous phases, respectively. This system is established by adding hydrophobic l-isopentyl tartrate (l-IPT) in organic strip phase (shell side) and hydrophilic sulfobutylether-β-cyclodextrin (SBE-β-CD) in aqueous feed phase (lumen side), which preferentially recognizes (+)-2R,4S-ketoconazole and (−)-2S,4R-ketoconazole, respectively. The studies performed involve two enantioselective extractions in a biphasic system, where KTZ enantiomers form four complexes with SBE-β-CD in aqueous phase and l-IPT in organic phase, respectively. The membrane is permeable to the KTZ enantiomers but non-permeable to the chiral selector molecules. Fractional chiral extraction theory, mass transfer performance of hollow fiber membrane, enantioselectivity and some experimental conditions are investigated to optimize the separation system. Mathematical model of I/II = 0.893e0.039NTU for racemic KTZ separation by hollow fiber extraction, is established. The optical purity for KTZ enantiomers is up to 90% when 9 hollow fiber membrane modules of 30 cm in length in series are used.
- I, (−)-2S,4R-ketoconazole;
- II, (+)-2R,4S-ketoconazole;
- CDs, cyclodextrin derivatives;
- l-IPT, l-isopentyl tartrate;
- d-IPT, d-isopentyl tartrate;
- HP-β-CD, hydroxypropyl-β-cyclodextrin;
- Me-β-CD, methyl-β-cyclodextrin;
- β-CD, β-cyclodextrin;
- NTU, number of transfer units;
- HTU, height of a transfer unit;
- PVDF,polyvinylidene fluoride
…………………….
Stereoselective synthesis of both enantiomers of ketoconazole from (R)- and (S)-
Stereoselective synthesis of both enantiomers of ketoconazole from (R)- and (S)-epichlorohydrin
Original Research Article- Pages 1283-1294
- Pelayo Camps, Xavier Farrés, Ma Luisa García, Joan Ginesta, Jaume Pascual, David Mauleón, Germano Carganico
- Bromobenzoates (2R,4R)- and (2S,4S)-18, prepared stereoselectively from (R)- and (S)-epichlorohydrin, were transformed into (2R,4S)-(+)- and (2S,4R)-(−)-Ketoconazole, respectively, following the known synthetic protocols for the racemic mixture.

Tetrahedron Asymmetry 1995, 6(6): 1283
Stereoselective syntheses of both enantiomers of ketoconazole (1) from commercially available (R)- or (S)-epichlorohydrin has been developed. The key-step of these syntheses involves the selective substitution of the methylene chlorine atom by benzoate on a mixture of  and
and  or of their enantiomers, followed by crystallization of the corresponding cis-benzoates, (2S,4R)-18 or(2S,4S)-18, from which (+)- or (−)-1 were obtained as described for (±)-1. The ee’s of (+)- and (−)-ketoconazole were determined by HPLC on the CSP Chiralcel OD-H.
or of their enantiomers, followed by crystallization of the corresponding cis-benzoates, (2S,4R)-18 or(2S,4S)-18, from which (+)- or (−)-1 were obtained as described for (±)-1. The ee’s of (+)- and (−)-ketoconazole were determined by HPLC on the CSP Chiralcel OD-H.
………………..
WO 1996029325
The incidence of fungal infections has considerably increased over the last decades. Notwithstanding the utility of the antifungal compounds commercialized in the last 15 years, the investigation in this field is however very extensive. During this time, compounds belonging to the azole class have beer, commercialized for both the topical and oral administrations, such a class including imidazoles as well as 1,2,4-triazoles. Some of these compounds car. show m some degree a low gastrointestinal tolerance as well as hepatotoxycity.
A large number of pharmaceutically active compounds are commercialized as stereoisomeric mixtures. On the other hand, the case in which only one of said stereoisomers is pharmaceutically active is frequent.
The undesired enantiomer has a lower activity and it sometimes may cause undesired side-effects.
Ketoconazole (1-acetyl-4-[4-[[2-(2,4-dichlorophenyl)-2-[(1H-imidazol-1-yl)methyl]-1,3-dioxolane-4-yl]methoxy]phenyl]piperazine), terconazole (1-[4-[[2(2,4-dichlorophenyl)-2-[(1H-1 , 2 ,4-triazol-1-yl)methyl]-1,3-dioxolane-4-yl]methoxy]phenyl]-4-(1-methylethyl)piperazine) and other related azole antifungal drugs contain in their structure a substituted 1,3-dioxolane ring, in which carbon atoms C2 and C4 are stereogenic centres, therefore four possible stereoisomers are possible. These compounds are commercialized in the form or cis racemates which show a higher antifungal activity than the corresponding trans racemates.
The cis homochiral compounds of the present invention, which are intermediates for the preparation of enantiomerically pure antifungal drugs, have been prepared previously in the racemic form and transformed into the different azole antifungal drugs in the racemic form [J. Heeres et al., J . Med . Chem . , 22 , 1003 (1979). J . Med . Chem . , 26, 611 (1983), J . Med . Chem . , 27 , 894 (1984) and US 4,144,346, 4,223,036, 4,358,449 and 4,335,125].
Scheme 1 shows the synthesis described for racemic ketoconazole [J. Heeres et al., J . Med . Chem . , 22 , 1003 (1979)]. Scheme 1
)

The synthesis of racemic terconazole [J. Heeres et al., J. Med . Chem . , 26 , 611 11983)] is similar. differing in the introduction of a 1 H- 1 , 2,4-triazol-1-yl substituent in place of 1H-imidazol-1-yl and in the nature of the phenol used in the last step of the synthetic sequence, which phenol is 1-methylethyl-4-(4- hydroxyphenyl)piperazme instead of 1-acetyl-4-(4-nydroxyphenyl)piperazine.
The preparation of racemic itraconazole [J. Heeres et al., J. Med . Chem. , 27 , 894 (1984)] is similar to that of terconazole, differing only in the nature of the phenol used in the last step of the synthetic sequence.

In the class of azoles containing a 1,3-dioxolane ring and a piperazine ring and moreover they are pure enantiomers, only the preparation of (+)- and (-)-ketoconazole has been described [D. M. Rotstein et al., J. Med . Chem . , 35, 2818 (1992)] (Scheme 2) starting from the tosylate of (+)- and (-) 2,2-dimethyl-1,3-dioxolane-4-methanol.
Scheme 2

This synthesis suffers from a series of drawbacks, namely: a) the use of expensive, high molecular weight starting products which are available only on a laboratory scale, and b) the need for several chromatographies during the process in order to obtain products of suitable purity, which maKes said synthesis economically unattractive and difficult to apply industrially.
Recently (N. M. Gray, WO 94/14447 and WO 94/14446) the use of (-)-ketoconazole and (+)-ketoconazole as antifungal drugs causing less side-effects than (±)-ketoconazole has been claimed.
The industrial preparation of enantiomerically pure antifungal drugs with a high antifungal activity and less side-effects is however a problem in therapy. The present invention provides novel homochiral compounds which are intermediates for the industrial preparation of already known, enantiomerically pure antifungal drugs such as ketoconazole enantiomers, or of others which have not yet been reported in literature, which are described first in the present invention, such as (+)-terconazole and (-)-terconazoie, which show the cited antifungal action, allowing to attain the same therapeutical effectiveness using lower dosages than those required for racemic terconazole
Example 14 : (2S,4R)-(-)-1-acetyl-4-[4-[ [2-(2,4-dichlorophenyl)-2-[(1H-imidazol-1-yl)-methyl]-1,3-dioxolane-4-yl]methoxy]phenyl]piperazine, (2S,4R) -(- )-ketoconazole.
This compound is prepared following the process described above for (2R,4S)-(+)-ketoconazole. Starting from HNa (60-65% dispersion in paraffin, 32 mg, 0.80 mmol), 1-acetyl-4-(4-hydroxyphenyl)piperazine (153 mg, 0.69 mol) and (2S,4S)-(-)-IV (Ar = 2,4-dichlorophenyl, Y = CH, R = CH3) (250 mg, 0.61 mmol), upon crystallization from an acetone:ethyl acetate mixture, (2S,4R) -(-)-ketoconazole is obtained [(2S,4R)-V Ar = 2,4-dichlorophenyl, Y = CH, Z = COCH3] (196 mg, 61% yield) as a solid, m.p. 153-155ºC (lit. 155-157ºC); [α]D 20 = -10.50 (c = 0.4, CHCl3) (lit. [α]D 25 = -10.58. c = 0.4, CHCl3) with e.e. > 99% (determined by HPLC using the chiral stationary phase CHIRALCEL OD-H and ethanol:hexane 1:1 mixtures containing 0.1 % diethylamine as the eluent).
+ KETOCONAZOLE…. UNDESIRED
Example 7: (2 R ,4S)-(+)-1-acetyl-4-[4-[[2-(2,4-dichlorophenyl)-2-[(1H-imidazol-1-yl)methyl]-1,3-dioxolane-4-yl]methoxy]phenyl]piperazine (22, 4 S)-(+)-ketoconazole.
To a suspension of NaH (dispersed in 60-65% paraffin, 19.2 mg, 0.48 mmol) in anhydrous DMSO (3 ml),
1-acetyl-4-(hydroxyphenyl)piperazine (102 mg, 0.46 mmol) is added and the mixture is stirred for 1 hour at room temperature. Then, a solution of (2R,4R) – (+)-IV (Ar = 2,4-dichlorophenyl, Y = CH, R = CH3) (160 mg, 0.39 mmol) in anhydrous DMSO (5 ml) is added, and the mixture is heated at 80ºC for 4 hours. The reaction mixture is allowed to cool to room temperature, diluted with water
(20 ml) and extracted with CH2Cl2 (3 × 25 ml). The combined organic phases are washed with water (3 × 25), dried with Na2SO4 and the solvent is evaporated off under vacuum. The oily residue thus obtained is crystallized from an acetone:ethyl acetate mixture to give (2R,4S)-(+)-ketoconazole ( (2R, 4 S) -V , Ar 2,4-dichlorophenyl, Y = CH , Z = COCH3 ) ( 110 mg , 5 3 % yie ld ) as a white solid, m.p. 155-156°C (lit. 154-156ºC), [α]D 20 = + 8.99 (c = 0.4, CHCl3) (lit. [α]D 25 = + 8.22, c = 0.4, CHCl3), with e.e. > 99% (determined by HPLC using the chirai stationary phase CHIRALCEL OD-H and ethanol:hexane 1:1 mixtures containing 0.1% of diethylamine, as the eluent; (+)-Ketoconazole retention time 73,28 min. (-)-Ketoconazole, retention time 79.06 min).
IR (KBr), ʋ : 2875, 1645, 1584, 1511, 1462, 1425, 1250, 103S, 313 cm-1.
1H NMR (500 MHz, CDCl3), δ : 2.12 (s, 3H, COCH3),
3.02 (m, 2H, 3-H2), 3.05 (m, 2H, 5-H2), 3.27 (dd, J= 9.5
Hz, J’=7.0 Hz, 1H) and 3.70 (dd, J=9.5 Hz, J’=5.0 Hz, 1 H) (4″-CH2), 3.60 (m, 2H, 6-H2), 3.76 (m, 2H, 2-H2), 3.73 (dd, J=8.0 Hz, J’=5.0 Hz, 1H) and 3.86 (dd, J=8.0 Hz, J’=6.5 Hz, 1H) (5″-H2), 4.34 (m, 1H, 4″-H), 4.40 (d, J=15.0 Hz, 1H) and 5.00 (d, J=15.0 Hz, 1H) (CH2-N), 4.34
(m, 1H, 4″-H), 6.76 [d, J = 9.0 Hz, 2H, 2'(C6' )-H], 6.88
[d, J=9.0 Hz, 2H, C3'(C5)-H], 6.96 (s, 1H, imidazole 5- H), 6.99 (s, 1H, imidazole 4-H), 7.25 (dd, J=8.5 Hz, J’=2.0 Hz, 1H, 5″‘-H), 7.46 (d, J=2.0 Hz, 1H, 3″‘-H),
7.53 (s, 1H, imidazole 2-H), 7.57 (d, J=8.5 Hz, 1H,
6″‘-H).
13C NMR (75.4 MHz, CDCI3), δ : 21.3 (CH3, COCH3), 41.4 (CH2, C2), 46.3 (CH2, C6), 50.6 (CH2, C3), 51.0 (CH2, C5), 51.2 (CH2, CH2-N), 67.6 [CH2, C5″ and 4″-CH2), 74.7 (CH, C4″), 108.0 (C, C2″), 115.2 [CH, C2'(6')], 118.8 [CH, C3'(5')], 121.2 (CH, imidazole C5), 127.2 (CH, C5″‘), 128.5 (CH, imidazole C4), 129.5 (CH, C6′”), 131.3 (CH, C3″‘), 133.0 (C, C2″‘), 134.6 (C, C1′”), 135.8 (C, C4″‘), 138.8 (CH, imidazole C2), 145.6 (C, C1′), 152.8 (C, C4′), 168.9 (C, CO).
…………………………
Experimental and theoretical analysis of the interaction of (+/-)-cis-ketoconazole with beta-cyclodextrin in the presence of (+)-L-tartaric acid
J Pharm Sci 1999, 88(6): 599
J Pharm Sci 1999, 88(6): 599
Enrico Redenti, Paolo Ventura, Giovanni Fronza, Antonio Selva, Silvia Rivara, Pier Vincenzo Plazzi and Marco Mor
Article first published online: 12 JUN 2000 | DOI: 10.1021/js980468o
1H NMR spectroscopy was used for determining the optical purity of cis-ketoconazole enantiomers obtained by fractional crystallization. The chiral analysis was carried out using β-cyclodextrin in the presence of (+)-l-tartaric acid. The mechanism of the chiral discrimination process, the stability of the complexes formed, and their structure in aqueous solution were also investigated by 1H and 13C chemical shift analysis, two-dimensional NOE experiments, relaxation time measurements, and mass spectrometry experiments. Theoretical models of the three-component interaction were built up on the basis of the available NMR data, by performing a conformational analysis on the relevant fragments on ketoconazole and docking studies on the components of the complex. The model derived from a folded conformation of ketoconazole turned out to be fully consistent with the molecular assembly found in aqueous solution, as inferred from NOE experiments. An explanation of the different association constants for the complexes of the two enantiomers is also provided on the basis of the interaction energies.
| WO1993019061A1 * | Mar 10, 1993 | Sep 30, 1993 | Janssen Pharmaceutica Nv | Itraconazole and saperconazole stereoisomers |
| WO1994025452A1 * | Apr 28, 1994 | Nov 10, 1994 | Ashit K Ganguly | Process for preparing intermediates for the synthesis of antifungal agents |
| EP0050298A2 * | Oct 13, 1981 | Apr 28, 1982 | Hoechst Aktiengesellschaft | 1-(1,3-Dioxolan-2-ylmethyl) azoles, process for their preparation and their use |
| EP0052905A1 * | Nov 19, 1981 | Jun 2, 1982 | Janssen Pharmaceutica N.V. | Novel (2-aryl-4-phenylthioalkyl-1,3-dioxolan-2-yl-methyl)azole derivatives |
| US5208331 * | Jun 18, 1992 | May 4, 1993 | Syntex (U.S.A.) Inc. | Process for preparing 1,3-dioxolane derivatives |
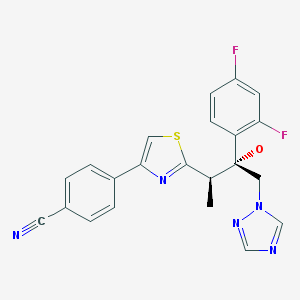
3 ISAVUCONAZOLE

 ISAVUCONAZOLE
ISAVUCONAZOLE



 ISAVUCONAZOLE
ISAVUCONAZOLE





Isavuconazole
4-{2-[(1R,2R)-(2,5-difluorophenyl)-2-hydroxy-1-methyl-3-(1H-1,2,4-triazol-1-yl)propyl]-1,3-thiazol-4-yl}benzonitrile
[(2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl)]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol;
(2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,4-difluorophenyl)-butan-2-ol
241479-67-4 CAS
946075-13-4 cas of SULPHATE (USAN) in phase 3Aspergillosis, Infection, candidal, RO-0098557
ROCHE Originator
30 September 2013
StockMarketWire.com – Basilea Pharmaceutica has revealed positive topline data from the isavuconazole phase 3 invasive aspergillosis study (SECURE). http://www.stockmarketwire.com/article/4677530/Basilea-reports-positive-results-from-study.htmlThe antifungal agent isavuconazole is being co-developed with Astellas Pharma Inc.The randomized, double-blind isavuconazole study (SECURE) achieved its primary objective in demonstrating non-inferiority versus voriconazole for the primary treatment of invasive fungal disease caused by Aspergillus species or certain other filamentous fungi.
Isavuconazole (BAL4815) is a triazole antifungal. Its prodrug, Isavuconazonium sulfate (BAL8557) is currently in two Phase IIIclinical trials (SECURE and VITAL), the results of which are expected in the second half of 2013.
On May 28, 2013, Basilea Pharmaceutica, the maker of the drug, announced it had been granted orphan drug status by the U.S.Food and Drug Administration (FDA).
BAL-4815, 241479-67-4, Bal4815, AC1OCFHQ, UNII-60UTO373KE, , BAL 4815, FT-0670421
Molecular Formula: C22H17F2N5OS Molecular Weight: 437.465086
Isavuconazium chloride hydrochloride (BAL-8557), a prodrug of Basilea Pharmaceutica’s BAL-4815 (isavuconazole), is a triazole in phase III clinical trials for the oral and intravenous treatment of severe fungal infections, including candidemia and other invasive Candida infections and invasive aspergillosis in immunocompromised patients. Additional phase III trials are ongoing for the treatment of invasive fungal infections caused by rare fungi. Phase II trials are ongoing for the treatment of candidal esophageal infection. Isavuconazole is water-soluble, highly bioavailable and can be administered in convenient once-daily or once-weekly dosing regimens.Originally developed at Roche, the drug candidate was subsequently acquired by Basilea. In May 2006, isavuconazium received fast track designation from the FDA for the treatment of infections caused by yeasts and molds, including fluconazole-resistant Candida strains, Aspergillus and zygomycetes in patients with weakened immune systems. In 2010, the product was licensed to Astellas Pharma by Basilea Pharmaceutica for codevelopment and copromotion worldwide, including an option for Japan, for the treatment of fungal infection. In 2013, FDA designated isavuconazium as a Qualified Infectious Disease Product (QIDP) designation for the treatment of invasive aspergillosis.
ISAVUCONAZOLE
CLINICAL TRIALS…LINK
PATENTS
| US8207352 | 6-27-2012 | Process for the manufacture of enantiomerically pure antifungal azoles as ravuconazole and isavuconazole |
| US2011281918 | 11-18-2011 | Antifungal Composition |
| US7803949 | 9-29-2010 | PROCESS FOR PREPARATION OF WATER-SOLUBLE AZOLE PRODRUGS |
| US7459561 | 12-3-2008 | N-substituted carbamoyloxyalkyl-azolium derivatives |
| US7189858 | 3-14-2007 | N-phenyl substituted carbamoyloxyalkyl-azolium derivatives |
| US6812238 | 11-3-2004 | N-substituted carbamoyloxyalkyl-azolium derivatives |
| US6300353 | 10-10-2001 | Azoles for treatment of fungal infections |
Several azoles are currently used for systemic mycoses. However, none of them fulfills the needs of clinical requirement in full extent, particularly with regard 0 to broad antifungal spectrum including aspergillus fumigatus, less drug-drug interaction, and appropriate plasma half-life for once a day treatment. Other clinical requirements which are not fulfilled by the azoles currently used, are efficacy against major systemic mycoses including disseminated aspergillosis, safety, and oral or parenteral formulations. Particularly, demand of a 5 parenteral administration of the azoles is increasing for the treatment of serious systemic mycoses. Most of the azoles on the market as well as under development are highly lipophilic molecules that make the parenteral formulation difficult.
Isavuconazole [(2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl)]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol; formula I, R1 and R3 represent fluorine and R2 represents hydrogen] as well as Ravuconazole [(2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl)]-1-(1H-1,2,4-triazol-1-yl)-2-(2,4-difluorophenyl)-butan-2-ol; formula I, R1 and R2 represent fluorine and R3 represents hydrogen] are useful antifungal drugs as reported in U.S. Pat. No. 5,648,372 from Feb. 1, 1995 or in U.S. Pat. No. 5,792,781 from Sep. 18, 1996 or in U.S. Pat. No. 6,300,353 from Oct. 9, 2001 (WO99/45008).
Since compounds of general formula I contain two adjacent chiral centers, synthesis of enantiomerically pure compound is complex and until now, all patented syntheses are not efficient enough and do not allow cost effective manufacturing on a technical scale:
Thus, U.S. Pat. Nos. 5,648,372 or 5,792,781 describe enantioselective synthesis of compounds of formula I (specifically Ravuconazole) from chiral 3-hydroxy-2-methyl propionic acid in 12 steps with overall yield lower than 5%. In another approach including 13 steps and low overall yield, (R)-lactic acid was used as the starting material (Chem. Pharm. Bull. 46(4), 623 (1998) and ibid. 46(7), 1125 (1998)).
Because both starting materials contain only one chiral center, in a number of inefficient steps, the second, adjacent chiral center has to be created by a diastereoselective reaction (using either Corey or Sharpless epoxidation method) which is not sufficiently selective leading mostly to a mixture of two diastereomers which have to be separated.
The second approach, based on (R)-methyl lactate, was recently very thoroughly optimized by BMS on a multi kilogram scale but it still does not fulfill requirements for cost effective manufacturing process (Organic Process Research & Development 13, 716 (2009)). The overall yield of this optimized 11 steps process is still only 16% (Scheme 1).

The manufacturing process for Isavuconazole is similar: Since Isavuconazole differentiates from Ravuconazole by only another fluorine substitution on the aromatic ring (2,5- instead of 2,4-difluorophenyl), the identical synthesis has been used (U.S. Pat. No. 6,300,353 from Oct. 9, 2001 and Bioorg. & Med. Chem. Lett. 13, 191 (2003)). Consequently, also this manufacturing process, based on (R)-lactic acid, faces the same problems: to many steps, extremely low overall yield and in addition to U.S. Pat. No. 6,300,353 claims even already known step as novel (claim 36).
Recent attempts to improve this concept as reported in WO 2007/062542 (Dec. 1, 2005), using less expensive, natural configured (S)-lactic acid, also failed: As already reported in U.S. Pat. No. 6,133,485 and in US 2003/0236419, the second chiral center was formed from an optically active allyl alcohol prepared in a few steps from (S)-lactic acid.
This allyl alcohol was subjected to Sharpless diastereoselective epoxidation providing first an opposite configured, epimeric epoxy alcohol which had to be then epimerized in an additional inversion step yielding finally the desired epoxy alcohol as the known precursor for Isavuconazole (U.S. Pat. No. 6,300,353). It is obvious that this process using less expensive (S)-lactic acid makes the entire process with an inversion step even more complex than the original approach.
Elegant and more efficient process has been claimed in US 2004/0176432 from Jun. 26, 2001) in which both chiral centers have been formed simultaneously, diastereo- and enantio-selectively pure in one single reaction step using chiral (R)-2-butynol as a chiral precursor in the presence of Pd(II)-catalyst and diethyl zinc (Scheme 2).

Since water soluble, (R)-2-butynol is expensive, recently identical process has been published, in which instead of (R)-2-butynol less water soluble and therefore, less expensive (R)-4-phenyl-3-butyn-2-ol was used (Synthetic Commun. 39, 1611 (2009)). Nevertheless, as incorrectly stated there, this process does not provide better diastereoselectivity than the original process using (R)-2-butynol: On the contrary disadvantage of this process is a very bad atom economy because huge phenyl group of (R)-4-phenyl-3-butyn-2-ol has to be “disposed” in oxidation step by the conversion of triple bond into carboxylic acid function.
All known processes for enantiomerically pure compounds of formula I have definitely too many operation steps and specifically very low overall yield. The chiral starting materials used, either 3-hydroxy-2-methyl propionic acid or (S)- or (R)-methyl lactate, contain only one chiral center and consequently, in number of steps, the second adjacent chiral center has to be ineffectively generated which makes the entire process long and expensive. The only known process, which generates both chiral centers simultaneously, requires again expensive chiral starting material (R)-2-butynol.
ISAVUCONAZOLE
…………………………………………….
synthetic scheme A, starting from 4-[(2R)-2-(3,4,5,6-tetrahydro-2H-pyran-2-yloxy)-propionyl]morpholine [which can be prepared by a same procedure as described in Chem. Pharm. Bull. 41, 1035, 1993.]. This synthesis route has been described for example in European Patent Application No. 99101360.8.


(a)
………………………………………………………………………
Example 1 (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,4-difluorophenyl)-butan-2-ol
To a solution of racemic 3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,4-difluorophenyl)-butan-2-ol (43.7 g) in acetone (800 ml) a solution of (1R)-10-camphorsulfonic acid (23 g) in methanol (300 ml) was added and the mixture was heated under reflux until a clear solution was obtained. The solution was slowly cooled to rt, seeded with crystals of the title enantiomeric salt and let overnight. The solid was collected by filtration, washed with acetone and dried to provide (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,4-difluorophenyl)-butan-2-ol (1R)-10-camphorsulfonate as white solid. This crude salt was then taken up in methylenechloride (100 ml) and water (ca. 100 ml) and the mixture was basified with aqueous sodium hydroxide solution. The organic layer was separated and the aqueous phase washed twice with methylenechloride (50 ml) and combined. The organic phases were then washed twice with water (2×50 ml), dried with sodium sulfate, filtrated and the solvent removed under reduced pressure. The crude product was then mixed with isopropanol (ca. 150 ml), heated for 10 min, cooled to 0° C. and stirred for ca. 2 hrs. The product was collected, washed with isopropanol and dried under reduced pressure to provide the enantiomerically pure title compound (17.5 g, 41% yield, 99.1% ee);
m.p. 164-166° C.; [α]=−30° (c=1, methanol, 25° C.);
NMR (CDCl3): 1.23 (3H, d, J=8 Hz), 4.09 (1H, q, J=8 Hz), 4.26 (1H, d, J=14 Hz), 4.92 (1H, d, J=14 Hz), 5.75 (1H, s), 6.75-6.85 (2H, m), 7.45-7.54 (2H, m), 7.62 (1H, s), 7.69 (1H, s), 7.75 (1H, d, J=8 Hz), 7.86 (1H, s), 8.03 (1H, d, J=8 Hz).
The analytical data were identical with published (U.S. Pat. No. 5,648,372 and Chem. Pharm. Bull. 1998, 46, 623-630).
Example 2 (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,4-difluorophenyl)-butan-2-ol
Racemic 3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,4-difluorophenyl)-butan-2-ol (44 g) and (1R)-10-camphorsulfonic acid (20 g) were suspended in methanol (ca. 300 ml), the slurry was stirred intensively, warmed up to ca. 70° C. and a small addition of acetic acid was added to obtain a clear solution. After cooling of the solution to rt and then to 0° C., the mixture was seeded with enantiomerically pure salt and stirred for another 2 hrs. The crystalline solid was collected by filtration, washed with cooled methanol and dried under reduced pressure. The crystals were partitioned between methylenechloride (300 ml) and saturated aqueous sodium bicarbonate solution (200 ml). The organic layer was washed twice with water (50 ml), dried with magnesium sulphate, filtrated and evaporated under reduced pressure to give the title compound (16.9 g, 38% yield, 95% ee). The analytical data were identical with published (U.S. Pat. No. 5,648,372 or Chem. Pharm. Bull. 1998, 46, 623).
Example 3 (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol
To a solution of racemic 3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol (10 g) in acetone (ca. 200 ml) a solution of (1R)-10-camphorsulfonic acid (3.9 g) in methanol (50 ml) was added and the mixture was heated shortly under reflux until a clear solution was obtained. The solution was then slowly cooled to rt, seeded with crystals of the desired enantiomeric salt and let overnight. The solid precipitate was collected by filtration, washed with acetone and dried to provide (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol (1R)-10-camphorsulfonate as white solid. This salt was then taken up in methylenechloride and water and basified with aqueous sodium bicarbonate solution. The organic layer was separated and the aqueous phase washed twice with methylenechloride. The organic phases were combined, dried with sodium sulphate, filtrated and the solvent removed under reduced pressure. The crude product was then dissolved in ethanol, the slurry heated for 20 min, small amount of water was added, the solution slowly cooled to 0° C. and stirred for ca. 2 hrs. The product was collected, washed with cold ethanol and dried under reduced pressure to provide the title enantiomerically pure compound (3.9 g, 39% yield, 96% ee). The analytical date were identical with published in U.S. Pat. No. 6,300,353 B1 and WO 99/45008.
Example 4 (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol
To a solution of racemic 3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol (100 g) in acetone (1000 ml) a solution of (1R)-10-camphorsulfonic acid (47 g) in methanol (500 ml) was added at rt, then slurry was heated under stirring to almost reflux for ca. 30 min, then cooled slowly to rt, seeded with the pure enantiomeric salt and stirred over night. The solid was collected by filtration, washed with methanol/acetone mixture, dried under reduced pressure. The residue was taken up with a solvent mixture of methylenechloride/water and after addition of saturated aqueous sodium bicarbonate solution the organic phase was separated and aqueous phase washed twice with methylenechloride. The combined organic phases were filtrated, the solvent removed under reduced pressure. Recrystallization of the crude product from aqueous ethanol provided enantiomerically pure title compound: 39 g (39% yield, 92% ee). The analytical data were identical with published: U.S. Pat. No. 6,300,353 and WO 99/45008.
Example 5 (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol
A solution of the racemic 3-[4-(4-cyanophenyl)thiazol-2-yl]-1-(1H-1,2,4-triazol-1-yl)-2-(2,5-difluorophenyl)-butan-2-ol (4.4 g) and (1R)-10-camphorsulfonic acid (2 g) in toluene (40 ml) containing glacial acetic acid (0.6 ml) was warmed up to approximately 70° C., then allowed to cool slowly to 20° C., seeded with the pure enantiomeric salt whereupon the pure enantiomeric salt start to crystallize out. After ca. 2 hrs at this temperature the solid was collected, washed with cold toluene and dried. The crystals were taken with a solvent mixture of methylenechloride/water and after addition of aqueous saturated sodium bicarbonate solution the organic phase was separated and aqueous phase washed twice with methylenechloride. The combined organic phases were filtrated and the solvent removed under reduced pressure. Recrystallization of the crude product from aqueous ethanol provided enantiomerically pure title compound: 2 g (45% yield, 99% ee). The analytical data were identical with published: U.S. Pat. No. 6,300,353 and WO 99/45008.
…………………………………..
WO 1999045008
The following synthetic scheme 1 illustrates the manufacture of one of the compounds of formula I′:

……………………………….
Bioorganic and medicinal chemistry letters, 2003 , vol. 13, 2 p. 191 – 196
A highly potent water soluble triazole antifungal prodrug, RO0098557 (1), has been identified from its parent, the novel antifungal agent RO0094815 (2). The prodrug includes a triazolium salt linked to an aminocarboxyl moiety, which undergoes enzymatic activation followed by spontaneous chemical degradation to release 2. Prodrug 1 showed high chemical stability and water solubility and exhibited strong antifungal activity against systemic candidiasis and aspergillosis as well as pulmonary aspergillosis in rats.
A highly potent water soluble triazole antifungal prodrug, RO0098557 (1), has been identified from its parent, the novel antifungal agent RO0094815 (2). The prodrug includes a triazolium salt linked to an aminocarboxyl moiety, which undergoes enzymatic activation followed by spontaneous chemical degradation to release 2. Prodrug 1 showed high chemical stability and water solubility and exhibited strong antifungal activity against systemic candidiasis and aspergillosis as well as pulmonary aspergillosis in rats.
- Scheme 1.
- Figure options
Chemistry
Scheme 1.We synthesized a series of new triazolium derivatives of Figure 1, Figure 3 and Scheme 1. CompoundsScheme 1 and Scheme 2, 6, 9, 10 and 11 were first prepared as outlined in Scheme 2 in order to analyze their stability and ability to release Figure 1, Figure 3 and Scheme 1. Next, aromatic analogues 18, 19, 20,21 and Figure 1, Figure 3 and Scheme 3 were synthesized for optimization of 11 to increase its water solubility and conversion rate. Compounds in the second series had sarcosine esters6 to make them water soluble, and they were also designed to generate acetaldehyde7 instead of formaldehyde for a better safety profile. The synthetic procedures for the second series of the derivatives are outlined in Scheme 3.- Scheme 2.
(a) ClCOOCH2Cl, diisopropylethylamine, CH2Cl2, rt (quant); (b) Figure 1, Figure 3 and Scheme 1, CH3CN, 80 °C (60%); (c) (1) ClCOOCH2Cl, Et3N, CH2Cl2, rt; (2) Ac2O, pyridine, rt (30%, two steps); (d) (1) NaI, CH3CN, 50 °C ; (2) Figure 1, Figure 3 and Scheme 1, CH3CN, 50 °C (88%, two steps); Synthesis of Scheme 1 and Scheme 2: (1) N-3-hydroxypropyl-N-methylamine, ClCOOCH2Cl, Et3N, CH2Cl2, rt; (2) AcCl, Et3N, CH2Cl2, rt (20%, two steps); (3) Figure 1, Figure 3 and Scheme 1, NaI, CH3CN, 50 °C (82%); Synthesis of 10: (1) l-prolinol, ClCOOCH2Cl, Et3N, CH2Cl2, rt; (2) Ac2O, pyridine, rt (<10%, 2 steps); (3) Figure 1, Figure 3 and Scheme 1, NaI, CH3CN, 50 °C (92%); Synthesis of 11: (1) 2-hydroxymethyl-N-methylaniline, ClCOOCH2Cl, diisopropylethylamine, CH2Cl2, rt; (2) Ac2O, diisopropylethylamine, rt (20%, two steps); (3)Figure 1, Figure 3 and Scheme 1, cat. NaI, CH3CN, reflux (63%). - Figure options
- Scheme 3.
(a) (1) oxalyl chloride, DMF, 0 °C; (2) KOtBu, THF, −5 °C (97%, two steps); (b) CH3NH2, MeOH, rt (90%); (c) LiAlH4, THF, 0 °C (80%); (d) (1) ClCOOCH(CH3)Cl, diisopropylethylamine, CH2Cl2, 0 °C; (2) Boc-Sarcosine, WSCI, DMAP, CH2Cl2, 0 °C (84%, two steps); (e) (1) Figure 1, Figure 3 and Scheme 1, NaI, CH3CN, 50 °C; (2) DOWEX-1 Cl− form, aqueous MeOH, rt (65%, two steps); (f) (1) HCl, EtOAc, rt; (2) lyophilization (69%, two steps); Synthesis of 18: (1) (i) (4,5-difluoro-2-methylaminophenyl)methanol, ClCOOCH(CH3)Cl, diisopropylethylamine, CH2Cl2, 0 °C; (ii) Boc-Sarcosine, WSCI, DMAP, CH2Cl2, 0 °C (quant, two steps); (2) Figure 1, Figure 3 and Scheme 1, cat. NaI, CH3CN, 80 °C; (50%,); (3) HCl, EtOAc, rt (90%); Synthesis of 19: (1) (i) 2-fluoro-6-methylaminophenyl)methanol, ClCOOCH(CH3)Cl, diisopropylethylamine, CH2Cl2, 0 °C; (ii) Boc-Sarcosine, WSCI, DMAP, CH2Cl2, 0 °C (74%, two steps); (2) Figure 1, Figure 3 and Scheme 1, cat. NaI, CH3CN, reflux; (3) HCl, EtOAc, rt (29%, two steps); Synthesis of 20: (1) (i) (5-fluoro-2-methylaminophenyl)methanol, ClCOOCH(CH3)Cl, diisopropylethylamine, CH2Cl2, 0 °C; (ii) Boc-Sarcosine, WSCI, DMAP, CH2Cl2, 0 °C (91%, two steps); (2) Figure 1, Figure 3 and Scheme 1, cat. NaI, CH3CN, 70 °C (72%); (3) HCl, EtOAc, rt (88%); Synthesis of 21: (1) (i) (4-chloro-2-methylaminophenyl)methanol, ClCOOCH(CH3)Cl, diisopropylethylamine, CH2Cl2, 0 °C; (ii) Boc-Sarcosine, WSCI, DMAP, CH2Cl2, 0 °C (71%, two steps); (2) Figure 1, Figure 3 and Scheme 1, CH3CN, 65 °C; (3) HCl, EtOAc, rt (65%, two steps).





read more at
Boyd, B.; Castaner, J. BAL-4815/BAL-8557
Drugs Fut 2006, 31(3): 187
Drugs Fut 2006, 31(3): 187
Antimicrobial Agents and Chemotherapy, 2008 , vol. 52, 4 p. 1396 – 1400
Ohwada, J.; Tsukazaki, M.; Hayase, T.; Oikawa, N.; Isshiki, Y.; Umeda, I.; Yamazaki, T.; Ichihara, S.; Shimma, N.Development of novel water antifungal, RO0098557
21st Med Chem Symp (November 28-30, Kyoto) 2001, Abst 1P-06
21st Med Chem Symp (November 28-30, Kyoto) 2001, Abst 1P-06
Ohwada, J.; Tsukazaki, M.; Hayase, T.; et al.
RO0098557, a novel water soluble azole prodrug for parenteral and oral administration (I). Design, synthesis, physicochemical properties and bioconversion42nd Intersci Conf Antimicrob Agents Chemother (ICAAC) (September 27-30, San Diego) 2002, Abst F-820
RO0098557, a novel water soluble azole prodrug for parenteral and oral administration (I). Design, synthesis, physicochemical properties and bioconversion42nd Intersci Conf Antimicrob Agents Chemother (ICAAC) (September 27-30, San Diego) 2002, Abst F-820
Tasaka et al., Chem. Pharm. Bull. 41(6) pp. 1035-1042 (1993).
| US4861879 | Feb 9, 1988 | Aug 29, 1989 | Janssen Pharmaceutica N.V. | [[4-[4-Phenyl-1-piperazinyl)phenoxymethyl]-1-3-dioxolan-2-yl]-methyl]-1H-imidazoles and 1H-1,2,4-triazoles |
| US5900486 | Sep 9, 1997 | May 4, 1999 | Hoffmann-La Roche Inc. | N-benzylazolium derivatives |
| AU4536497A | Title not available | |||
| EP0667346A2 | Feb 3, 1995 | Aug 16, 1995 | Eisai Co., Ltd. | Azole antifungal agents, process for the preparation there of and intermediates |
| WO1992017474A1 | Mar 26, 1992 | Oct 15, 1992 | Pfizer | Triazole antifungal agents |
| US5648372 | Feb 1, 1995 | Jul 15, 1997 | Eisai Co., Ltd. | Antifungal agents, and compositions |
| US5686646 * | May 23, 1995 | Nov 11, 1997 | Schering-Plough Corporation | Chiral hydrazine derivatives |
| US5746840 * | Mar 28, 1997 | May 5, 1998 | Janssen Pharmaceutica, N.V. | Process for preparing enantiomerically pure 6-{4-chlorophenyl) (1 H-1,2,4-triazol-1-YL) methyl}-1-methyl-1 H-benzotriazole |
| US5792781 | Sep 18, 1996 | Aug 11, 1998 | Eisai Co., Ltd. | Antifungal agents, processes for the preparation thereof, and intermediates |
| US6020497 | Oct 9, 1998 | Feb 1, 2000 | Merck & Co., Inc. | 3-substitutes isoxazolidines as chiral auxiliary agents |
| US6133485 | Apr 15, 1998 | Oct 17, 2000 | Synphar Laboratories, Inc. | Asymmetric synthesis of 2-(2,4-difluorophenyl)-1-heterocycl-1-yl butan-2,3-diols |
| US6300353 | Mar 5, 1999 | Oct 9, 2001 | Basilea Pharmaceutica Ag, A Swiss Company | Azoles for treatment of fungal infections |
| US6383233 | Mar 7, 1997 | May 7, 2002 | Reuter Chemicscher Apparatebau Kg | Separation process |
| US6812238 * | Oct 31, 2000 | Nov 2, 2004 | Basilea Pharmaceutica Ag | N-substituted carbamoyloxyalkyl-azolium derivatives |
| US7151182 * | Sep 3, 2004 | Dec 19, 2006 | Basilea Pharmaceutica Ag | Intermediates for N-substituted carbamoyloxyalkyl-azolium derivatives |
| US7803949 * | Dec 20, 2006 | Sep 28, 2010 | Eisai R&D Management Co., Ltd. | Process for preparation of water-soluble azole prodrugs |
| US20030236419 | Dec 31, 2002 | Dec 25, 2003 | Sumika Fine Chemicals Co., Ltd. | Production methods of epoxytriazole derivative and intermediate therefor |
| US20040176432 | Jun 17, 2002 | Sep 9, 2004 | Milan Soukup | Intermediate halophenyl derivatives and their use in a process for preparing azole derivatives |
| WO2003002498A1 * | Jun 17, 2002 | Jan 9, 2003 | Basilea Pharmaceutica Ag | Intermediate halophenyl derivatives and their use in a process for preparing azole derivatives |
4
TIOCONAZOLE
TIOCONAZOLE

Tioconazole;UK-20349;Trosyd;Trosyl;Vagistat-1
l-[2-(2-chloro-3-thienyl)methoxy]-2-(2,4- dichlorophenyl)ethyl]-lH-imidazole,
1-[2-(2-Chloro-3-thienylmethoxy)-2-(2,4-dichlorophenyl)ethyl]-1H-imidazole
65899-73-2
Launched – 1983, Bristol-Myers Squibb
Trademarks: Fungibacid (Asche); Gyno-Trosyd (Pfizer); Trosyd (Pfizer); Trosyl (Pfizer); Vagistat (BMS); Zoniden (Irbi)
Molecular Formula: C16H13Cl3N2OS
Molecular Weight: 387.71
Percent Composition: C 49.57%, H 3.38%, Cl 27.43%, N 7.23%, O 4.13%, S 8.27%
Derivative Type: Hydrochloride
Molecular Formula: C16H13Cl3N2OS.HCl
Molecular Weight: 424.17
Percent Composition: C 45.31%, H 3.33%, Cl 33.43%, N 6.60%, O 3.77%, S 7.56%
Properties: Crystals, mp 168-170°.
Melting point: mp 168-170°
Therap-Cat: Antifungal (topical).
Tioconazole is an antifungal medication of the imidazole class used to treat infections caused by a fungus or yeast. It is marketed under the brand names Trosyd and Gyno-Trosyd (Pfizer). Tioconazole ointments serve to treat women’s vaginal yeast infections.[1]They are available in one day doses, as opposed to the 7-day treatments more common in use in the past.
Tioconazole topical (skin) preparations are also available for ringworm, jock itch, athlete’s foot, and tinea versicolor or “sun fungus”.
Side effects
Side effects (for the women’s formulas) may include temporary burning/irritation of the vaginal area, moderate drowsiness, headachesimilar to a sinus headache, hives, and upper respiratory infection. These side effects may be only temporary, and do not normally interfere with the patient’s comfort enough to outweigh the end result.
| |
| SYSTEMATIC (IUPAC) NAME | |
|---|---|
| (RS)-1-[2-[(2-Chloro-3-thienyl)methoxy]-2-(2,4-dichlorophenyl)ethyl]-1H-imidazole | |
| CLINICAL DATA | |
| TRADE NAMES | Vagistat-1 |
| AHFS/DRUGS.COM | monograph |
| LEGAL STATUS | |
| ROUTES | Topical |
| IDENTIFIERS | |
| CAS NUMBER | 65899-73-2 |
| ATC CODE | D01AC07 G01AF08 |
| PUBCHEM | CID 5482 |
| DRUGBANK | DB01007 |
| KEGG | D00890 |
| SYNONYMS | Thioconazole |
| CHEMICAL DATA | |
| FORMULA | C16H13Cl3N2OS |
| MOL. MASS | 387.711 g/mol |
Imidazole derivatives, in particular, l-[2-(2-chloro-3-thienyl)methoxy]-2-(2,4- dichlorophenyl)ethyl]-lH-imidazole, commonly referred to as tioconazole, are known for their antifungal therapeutic properties. US 4,062,966 discloses a process for the preparation of l-aryl-2-(l -imidazolyl) alkyl ethers and thioethers which employs arylation of an appropriate 1 -aryl-2-(l -imidazolyl)alkanol or alkane thiol having the formula

wherein Rl to R4 are each H or C,^ alkyl, Ar is phenyl, or substituted phenyl wherein said substitutents are halogen, C,^ alkyl, C,_6 alkoxy, thienyl, or halothienyl, and, Z is oxygen or sulfur. In accordance with US’966, the reaction comprises converting the alcohol or thiol in a suitable solvent to its alkali metal derivative by treatment with a strong base, such as an alkali metal amide or hydride, and reacting with the appropriate aralkyl halide ofthe formula
X-(CH2)η-Y
where n is 1 or 2, Y is an aromatic heterocyclic group or substituted heterocyclic group, wherein substitutents are halogen, C,.6 alkyl, or C,.6 alkoxy atoms, thienyl or halothienyl group, and X is a halogen, preferably chlorine. Tetrahydrofuran (THF) is the preferred solvent taught in US ‘966. Reaction temperatures may range from about 0 °C to reflux temperature ofthe solvent and reaction times range from about 1 hour to about 24 hours. The product is isolated with water, extracted with ether, and may be purified as the free base or converted to a salt, e.g. the hydrochloride, and purified by recrystallization. A disadvantage ofthe process disclosed in US ‘966 is that THF is a peroxide generator which presents the potential for an explosion. From a commercial viewpoint, peroxide generators are not preferred due to the dangers associated therewith.
GB 1 522 848 discloses a process for the preparation of imidazoles useful as antifungal agents involving a labor intensive, multi-sequence reaction of an imidazole ether with a reactive ester. Like US ‘966, THF is employed presenting similar concerns in the synthesis ofthe desired imidazole product.
According to the Pharmaceutical Manufacturing Encyclopedia, tioconazole is prepared by dissolving l-(2,4-dichlorophenyl)-2-(l- imidazolyl)ethanol in THF and sodium hydride and heating to about 70 βC. The resulting mixture is then contacted with 2-chloro-3- chloromethylthiophene and heated to reflux (about 67 CC). The resulting product is filtered, saturated with hydrogen chloride, triturated and recrystallized to obtain the purified tioconazole hydrochloride product having a melting point of about 170 βC. This salt must then be freebased to form the product used in pharmaceutical formulations. This route, like those discussed above, also presents the dangers of a potential explosion. There is thus a continuing need for a commercially viable, synthetic route for the production of imidazoles, in particular tioconazole.
…………………….
see US 4062966
………………………….
References
- Tioconazole, Mayo Clinic
- References1:Gymer, G.E.; DE 2619381 .References2:Hillier, K.; Blancafort, P.; Castaner, J.; Serradell, M.N.; Tioconazole. Drugs Fut 1980, 5, 10, 509.
- Growth quantification and rapid drug susceptibility testing of uropathogenic Candida albicans by isothermal microcalorimetry
28th Congr Eur Assoc Urol (March 15-19, Milan) 2013, Abst 618 - Difference in percutaneous absorption and intracutaneous distribution in guinea pigs among topical antifungal drugs (tioconazole solution, tioconazole cream, miconazole nitrate solution and bifonazole solution)
Biol Pharm Bull 2004, 27(9): 1428 - A randomized comparison of the nail surface remainder of three nail lacquers containing amorolfine 5%, ciclopirox 8%, or tioconazole 28% in healthy volunteers
63rd Annu Meet Am Acad Dermatol (AAD) (February 18-22, New Orleans) 2005, Abst P1805
Literature References:
Antimycotic imidazole derivative. Prepn: G. E. Gymer, BE 841309; idem, (1976, 1977 both to Pfizer).
Antifungal spectrum: S. Jevons, Antimicrob. Agents Chemother. 15, 597 (1979); F. C. Odds, J. Antimicrob. Chemother. 6,749 (1980).
Pharmacology: M. S. Marriott et al., Dermatologica 166, Suppl. 1, 1 (l983).
Clinical trial in dermatomycosis: Y. M. Clayton et al., Clin. Exp. Dermatol. 7, 543 (1982). Series of articles on pharmacology and clinical efficacy in gynecological use:Gynak. Rundsch. 23, Suppl. 1, 1-60 (l983).
5 FOSRAVUCONAZOLE

Fosravuconazole
Phosphoric acid 2(R)-[4-(4-cyanophenyl)thiazol-2-yl]-1(R)-(2,4-difluorophenyl)-1-(1H-1,2,4-triazol-1-ylmethyl)propyoxymethyl monoester
(2R,3R)-3-r4-(4-cyanophenyl)thiazol-2-yll-2-(2,4-difluorophenyl)- 1 -(1 H- 1 ,2,4- triazol-l-yl)-2-[(dihydrogen phosphonoxy)methoxylbutane
BEF-1224
BMS-379224
E-1224
BMS-379224
E-1224
Phosphoric acid 2(R)-[4-(4-cyanophenyl)thiazol-2-yl]-1(R)-(2,4-difluorophenyl)-1-(1H-1,2,4-triazol-1-ylmethyl)propyoxymethyl monoester bis(L-lysine) salt is used as drug
The azole antifungal agent E-1224 is a prodrug of ravuconazole. In 2009, originator Eisai licensed E-1224 to Drugs for Neglected Diseases Initiative for the treatment of American trypanosomiasis (Chagas disease) in Latin America and the Caribbean. DNDi was conducting phase II clinical trials with the prodrug for this indication, however, development of the compound has been discontinued due to lack of sustained efficacy. Ravuconazole was originally licensed by Eisai to Bristol-Myers Squibb (BMS). BMS developed the drug’s prodrug, referred to by BMS as BMS-379224. For strategic reasons, BMS did not pursue development of the compound. In 2010, E-1224 was licensed exclusively to Brain Factory for development, commercialization and sublicense in Japan for the treatment of fungal infections.
About Ravuconazole and Ravuconazole Prodrug
The compound on the left is ravuconazole; the compound on the right is the dihydrogen phosphonoxy methoxy derived ravuconazole prodrug which has improved solubility and bioavailability.

The compound on the left is ravuconazole; the compound on the right is the dihydrogen phosphonoxy methoxy derived ravuconazole prodrug which has improved solubility and bioavailability.
……………………………………………………………
WO 2001052852
Triazole antifungal compounds are well known in the prior art. Of the several classes known, one particularly potent class contains a tertiary hydroxyl group. For example, U. S. Patent 5,648,372 discloses that (2R,3R)-3-[4-(4- cyanophenyl)thiazol-2-yl]-2-(2,4-difluorophenyl)- 1 -( 1 H- 1 ,2,4-triazol- 1 -yl)- butan-2-ol has anti-fungal activity.

The utility of this class of compounds is limited by their low water solubility. For example, the solubility of the above triazole compound in water at pH 6.8 is 0.0006 mg/mL. This greatly impedes developing suitable parenteral dosage forms.
One method of addressing this problem was disclosed in European Patent Application 829478, where the water solubility of an azole antifungal agent was increased by attaching a linked amino-acid to the azole portion of the molecule

Alternatively, WO 97/28169 discloses that a phosphate moiety can be attached directly to the tertiary hydroxyl portion of the anti-fungal compound, e.g. the compound having the formula

U.S. Patent 5,707,977 and WO 95/19983 disclose water soluble prodrugs having the general formula

wherein X is OP(O)(OH)2 or an easily hydrolyzable ester OC(O)RNR l’rR>2.
WO 95/17407 discloses water-soluble azole prodrugs of the general formula

wherein X is P(O)(OH)2, C(O)-(CHR’)n-OP(O)(OH)2 or C(O)-(CHR’)π
-(OCHR,CHR1)mOR2.
WO 96/38443 discloses water-soluble azole prodrugs of the general formula

U.S. Patent 5,883,097 discloses water-soluble amino acid azole prodrugs such as the glycine ester

The introduction of the phosphonooxymethyl moiety into hydroxyl containing drugs has been disclosed as a method to prepare water-soluble prodrugs of hydroxyl containing drugs.
European Patent Application 604910 discloses phosphonooxymethyl taxane derivatives of the general formula

wherein at least one of R1 ‘, R2″, R3′, R6′ or R7′ is OCH2OP(O)(OH)2.
European Patent Application 639577 discloses phosphonooxymethyl taxane derivatives of the formula T-[OCH2(OCH2)mOP(O)(OH)2]n wherein T is a taxane moiety bearing on the C13 carbon atom a substituted 3-amino-2- hydroxypropanoyloxy group; n is 1, 2 or 3; m is 0 or an integer from 1 to 6 inclusive, and pharmaceutically acceptable salts thereof. WO 99/38873 discloses O-phosphonooxymethyl ether prodrugs of a diaryl 1,3,4-oxadiazolone potassium channel opener.
Golik, J. et al, Bioorganic & Medicinal Chemistry Letters, 1996, 6:1837- 1842 discloses novel water soluble prodrugs of paclitaxel such as


EXAMPLE 1
(2R,3R)-3-r4-(4-cyanophenyl)thiazol-2-yll-2-(2,4-difluorophenyl)- 1 -(1 H- 1 ,2,4- triazol-l-yl)-2-[(dihydrogen phosphonoxy)methoxylbutane, sodium salt

(2R,3R)-3-r4-(4-cyanophenyl)thiazol-2-yll-2-(2,4-difluorophenyl)-l-(lH- 1 ,2,4-triazol- 1 -yl)-2-[(di-tert-butyl phosphonoxy)methoxy1butane

To a solution of (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-2-(2,4- difluorophenyl)-l-(lH-l,2,4-triazol-l-yl)butan-2-ol, II, (8.74 g, 20 mmol) in THF (40 mL) under a nitrogen atmosphere was added sodium hydride (0.80 g, 60% in oil, 20 mmol) at rt. The resulting mixture was stirred at rt for 0.25 h and then di- tert-butyl chloromethyl phosphate, III (10.3 g, 40 mmol) was added. The reaction mixture was heated at 50 °C for 16 h. The reaction mixture was then allowed to cool to rt and was concentrated under reduced pressure. The residue was dissolved in Et2O and was washed with H2O and brine. The organic layer was dried over MgSO4 and was concentrated under reduced pressure to obtain 17.0 g of crude subtitled compound. IV, as a gum. A small portion of this crude compound was purified by reverse phase chromatography on C- 18. The column was eluted with 30% CH3CN/H2O, 38% CH3CN/H2O, 45% CH3CN/H2O and then 50% CH3CN/Η2O. The product containing fractions were concentrated under reduced pressure in order to remove CH3CN. The resulting aqueous layer was then extracted with Et2O. The Et O layers were washed with brine, dried and concentrated under reduced pressure to afford purified subtitled compound, IV, as a white solid. 1H NMR (300 MHz, CDC13): δ 8.35 (s, 1H), 7.98 (d, 2H, J=9), 7.76 (s, 1H), 7.71 (d, 2H, J=9), 7.63 (s, 1H), 7.36-7.27 (m, 1H), 6.86-6.78 (m, 2H), 5.53 (dd, 1H, J=28,6), 5.53 (dd, 1H, J=9,6), 5.17 (d, 1H, J=15), 5.03 (d, 1H, J=15), 4.01 (q, 1H, J=7), 1.47 (s, 9H), 1.45 (s, 9H), 1.37 (d, 3H, J=7). MS [ESI+ (M+H)+] 660.2 obs. B. (2R,3R)-3-r4-(4-cyanoρhenyl)thiazol-2-yll-2-(2,4-difluorophenyl)-l-(lH- 1 ,2,4-triazol-l-yl)-2-[(dihydrogen phosphonoxy)methoxy]butane, sodium saltdeprotection


The crude (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-2-(2,4- difluoropheny 1)- 1 -( 1 H- 1 ,2 ,4-triazol- 1 -y l)-2- [(di-tert-buty 1 phosphonoxy)methoxy]butane, IV, (17 g) was dissolved in CH C1 (100 mL). To this solution was added TFA (50 mL) and the reaction mixture was stirred at rt for 0.25 h. The reaction mixture was then concentrated under reduced pressure. To the residue was added H2O (200 mL), Et2O (100 mL) and EtOAc (100 mL). The pH of the aqueous layer was adjusted to 7.6 by addition of solid Na2CO3 and then the organic and aqueous layers were separated. The aqueous layer was then subjected to reverse phase chromatography on 400 g of C-18 eluted with H2O to 5% CH3CN/Η2O. The product containing fractions were concentrated under reduced pressure, frozen and lyophilized to afford 1.5 g of the subtitled compound, I, as a white solid. (1.5 g, 12% over two steps). Η NMR (500 MHz, D2O) δ 8.91 (s, IH), 7.92 (s, IH), 7.81 (d, 2H, J=8), 7.80 (s, IH), 7.77 (d, 2H, J=8), 7.21 (dd, IH, J=15,9), 6.99 (ddd, IH, J=9,9,2), 6.91 (ddd, IH, J=9,9,2), 5.35 (dd, IH, J=6,6), 5.29 (d, IH, J=15), 5.21 (dd, IH, J=6,6), 5.19 (d, IH, J=15), 3.86 (q, IH, J=7), and 1.35 (d, 3H, J=7); MS [(ESI" (M-HV 546.1]; Anal. Calcd for C23Hi8F2N5θ5SιPι Na2/3.5 H2O: C, 42.21 : H, 3.85: N, 10.70: Na, 7.03. Found: C, 42.32: H, 3.83: N, 10.60: Na, 7.04.
Di-tert-butyl chloromethyl phosphate, III:
Di-tert-butyl chloromethyl phosphate, III, may be made by any of the following methods.
Method 1
Silver di-t-butyl phosphate (6.34 g, 20 mmol), which was prepared by mixing di- t-butyl phosphate (obtained from di-t-butyl phosphite by the method of Zwierzak and Kluba, Tetrahedron, 1971 , 27, 3163) with one equivalent of silver carbonate in 50% aqueous acetonitrile and by lyophilizing to dryness, was placed together with chloroiodomethane (35 g, 200 mmol) in benzene and stirred at room temperature for 18 hrs. The reaction mixture was filtered and the filtrate concentrated under reduced pressure. The residue was chromatographed on silica and eluted with 2:1 hexanes-ethyl acetate. Appropriate fractions were concentrated to dryness to obtain the subtitled compound III (3.7 g, 71% yield): H NMR (CDCI3) δ 5.63 (d, 2H, J=17), 1.51 (s, 18H); MS (MH+ = 259).
Method 2
Tetrabutylammonium di-t-butyl phosphate was prepared by dissolving di-t-butyl phosphate [ 20g, 94 mmol (obtained from di-t-butyl phosphite by the method of Zwierzak and Kluba, Tetrahedron, 1971, 27, 3163)] in methanolic tetrabutylammonium hydroxide (47 mL of 1M solution, 47 mmol). The reaction mixture had a temperature of 23 °C and pH of 4.33. The pH of the reaction mixture was adjusted to 6.5-7.0 by addition of methanolic tetrabutylammonium hydroxide (48 mL of 1M solution, 48 mmol) over 0.2 h. The reaction mixture was stirred for 0.5 h at approximately 26 °C and then was concentrated under reduced pressure at a bath temperature below 40 °C. The crude residue was azeotroped three times by adding toluene (3×100 mL) and then the mixture was concentrated under reduced pressure. The crude residue was then triturated in cold hexanes (0°C) for 1 h and then the solid was collected by filtration, washed with a minimum amount of cold hexanes and dried to give a first crop of tetrabutylammonium di-t-butyl phosphate as a white solid. (24. Og). The mother liquor was concentrated under reduced pressure and then triturated in cold hexanes (20 mL) for 1 h. The solid was collected by filtration, washed with a minimum amount of cold hexanes and dried to give a second crop of tetrabutylammonium di-t-butyl phosphate as a white solid. [(8.5g), 32.5g total (77%)]. A solution of tetrabutylammonium di-t-butyl phosphate (218 g, 480 mmol) in benzene (200 mL) was added dropwise to stirred chloroiodomethane (800g, 4535 mmol) over 1.5 h at rt. The reaction mixture was stirred an additional 1.5 h at rt and then was concentrated under reduced pressure. The oily residue was dissolved in Et2O and filtered to remove white solids which had precipitated. The organic layer was washed with saturated NaHCO3 and H O/brine (1/1). The organic layer was then dried over magnesium sulfate, filtered and concentrated under reduced pressure to yield a red brown oil (320 g). The red brown oil was subjected to chromatography on silica gel (800g) eluted with 20% EtOAc/Hexanes, 25% EtOAc/Hexanes then 30% EtOAc/Hexanes. The product containing fractions were concentrated under reduced pressure to yield a golden oil. The oil was diluted with CH2C12 (30 mL) , concentrated under reduced pressure and then dried under vacuum to yield the subtitled compound III (61.3g, 49% yield). 1H NMR (Benzene-d6) δ 5.20 (2H, d, J=15), 1.22 (18H, s).
Method 3
Iodochloromethane (974 g, 402 mL, 5.53 mol) at 25°C was treated with tetrabutylammonium di-t-butylphosphate (250 g, 0.553 mol). The phosphate was added portion wise over 10 minutes. The heterogeneous mixture became a clear pink solution after approximately 15 minutes. The mixture was stirred for three hours, and the iodochloromethane was then removed by rotary evaporation with a bath temperature of <30°C. The residue was taken up in 1 L t-butyl methyl ether and stirred for 15 minutes to precipitate tetrabutylammonium iodide by-product. Tetrabutylammonium iodide was removed by vacuum filtration through a sintered glass funnel. The filtrate was concentrated by rotary evaporation to an oil which contained a 5:1 mixture of III and undesired dimer impurity

III”
The mixture can be purified by a silica gel chromatography to obtain III as pure compound in ~60% yield as an oil.
EXAMPLE 2
(2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-2-(2,4-difluorophenyl)-l-(lH-l,2,4- triazol- 1 -yl)-2- (dihydrogen phosphonoxy)methoxy]butane
A. An oven dried, 1L round-bottom flask equipped with a mechanical stirrer, nitrogen inlet adapter, pressure-equalizing addition funnel fitted with a rubber septum and temperature probe was charged with sodium hydride (2.89 g, 0.069 mol, 60%) and THF (50 mL). To this stirred suspension, (2R,3R)-3-[4-(4- cyanophenyl)thiazol-2-yl]-2-(2,4-difluorophenyl)- 1 -( 1 H- 1 ,2,4-triazol- 1 -yl)butan- 2-ol, II, (10 g, 0.023 mol) in 30 mL of THF was added dropwise over 20 minutes at room temperature. After stirring for 45 minutes, a solution of iodine (2.99 g, 0.0115 mol) in THF (30 mL)) was added dropwise over 10 minutes followed by dropwise addition of compound di tert butylchloromethyl phosphate, III (13.29 g, 0.035 mol, -68% purity) over 15 minutes. The reaction mixture was stirred for 4 hours at about 41 °C to complete the reaction. The completion of the reaction was judged by in-process HPLC. The reaction mixture was poured into ice cold water (100 mL). The aqueous phase was separated and extracted with ethyl acetate (3 x 50 mL) and the combined organic extract was washed with 10% sodium thiosulfite (50 mL), water (50 mL), brine (50 mL), dried over magnesium sulfate and concentrated under reduced pressure to give pale yellow oil (22.8 g, In-process HPLC: ~ 97% pure). The crude product was used “as is” in step B.
B. To a round-bottom flask equipped with magnetic stirrer, cooling bath, pH probe and N2 inlet-outlet was charged the product of Step A above (7.5 g) in CH2C12 (23 mL) and cooled to 0 °C. To this stirred solution, trifluoroacetic acid (8.8 mL) was added slowly and stirred for 3 h to complete the reaction. The completion of the reaction was judged by in-process HPLC. The reaction mixture was poured into a cold solution of 2N NaOH (64 mL). The reaction mixture was extracted with t-butyl acetate (2 x 65 mL) to remove all the organic impurities. The aqueous layer containing the title product as bis sodium salt was treated with activated charcoal (10 g) and filtered through a bed of Celite. The clear filtrate was acidified with IN HC1 to pH 2.5. The free acid, the title product, was extracted into ethyl acetate (2 x 50 mL). The combined organic layer was washed with water, dried over MgSO4) filtered, and the filtrate concentrated under reduced pressure to afford 3.39 g of crude title product.
EXAMPLE 3
Bis lysine salt of (2R,3R)-3-r4-(4-cyanophenyl)thiazol-2-yl]-2-(2,4- difluorophenyl)- 1 -( 1 H- 1 ,2,4-triazol- 1 -yl)-2-[(dihydrogen phosphonoxy)methoxy]butane
The above obtained title product from Example 2 was dissolved in methanol (75 mL) and to this L-lysine (1.8 g) was added and heated at 60 °C for 4.5 h. The hot reaction mixture was filtered through a bed of Celite. The filtrate was concentrated to about 5 mL, mixed with ethanol (100 mL) and heated to 65 °C to crystallize the bis lysine salt. The salt was collected on a Buchner funnel and dried under vacuum to afford 3.71 g of the title compound as an off white crystalline solid.
About Eisai Co., Ltd.
Eisai Co., Ltd. is a research-based human health care (hhc) company that discovers, develops, and markets products throughout the world. Eisai focuses its efforts in three therapeutic areas: integrative neuroscience, including neurology and psychiatric medicines; integrative oncology, which encompasses oncotherapy and supportive-care treatments; and vascular and immunological reactions. Eisai contributes to the well-being of people around the world through a global network of research facilities, manufacturing sites and marketing subsidiaries. For more information about Eisai Co., Ltd., please visithttp://www.eisai.co.jp/index-e.html.
Eisai Co., Ltd. is a research-based human health care (hhc) company that discovers, develops, and markets products throughout the world. Eisai focuses its efforts in three therapeutic areas: integrative neuroscience, including neurology and psychiatric medicines; integrative oncology, which encompasses oncotherapy and supportive-care treatments; and vascular and immunological reactions. Eisai contributes to the well-being of people around the world through a global network of research facilities, manufacturing sites and marketing subsidiaries. For more information about Eisai Co., Ltd., please visithttp://www.eisai.co.jp/index-e.html.
ref
BMS-379224, a water-soluble prodrug of ravuconazole
42nd Intersci Conf Antimicrob Agents Chemother (ICAAC) (September 27-30, San Diego) 2002, Abst F-817
42nd Intersci Conf Antimicrob Agents Chemother (ICAAC) (September 27-30, San Diego) 2002, Abst F-817
| WO2000030655A1* | Nov 17, 1999 | Jun 2, 2000 | Squibb Bristol Myers Co | Water soluble prodrugs of azole compounds |
| WO2006118351A1 | May 1, 2006 | Nov 9, 2006 | Eisai Co Ltd | Mono-lysine salts of azole compounds |
| WO2012060448A1 | Nov 4, 2011 | May 10, 2012 | Eisai R&D Management Co., Ltd. | Combined pharmaceutical composition as antifungal agent |
| CN101341160B | Dec 20, 2006 | Jan 25, 2012 | 卫材R&D管理有限公司 | Process for production of water-soluble azole prodrug |
| EP1345915A1 * | Oct 18, 2001 | Sep 24, 2003 | Bristol-Myers Squibb Company | Improved process for water soluble azole compounds |
| EP2291084A1 * | May 20, 2009 | Mar 9, 2011 | Neurogesx, Inc. | Carbonate prodrugs and methods of using the same |
| US7230023 | Aug 20, 2003 | Jun 12, 2007 | Sankyo Company, Limited | Triazole compound containing a phosphonate group |
| US8735376 | May 20, 2009 | May 27, 2014 | Acorda Therapeutics, Inc. | Carbonate prodrugs and methods of using the same |
6 RAVUCONAZOLE
BMS-207147, ER-30346
- BMS 207147
- ER 30346
- Ravuconazole
- UNII-95YH599JWV
CAS Registry Number: 182760-06-1
CAS Name: 4-[2-[(1R,2R)-2-(2,4-Difluorophenyl)-2-hydroxy-1-methyl-3-(1H-1,2,4-triazol-1-yl)propyl]-4-thiazolyl]benzonitrile
Additional Names: (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-2-(2,4-difluorophenyl)-1-(1H-1,2,4-triazol-1-yl)-2-butanol
(2R,3R)-3-i4-(4-cvanophenyl)thiazol-2-yl1-1 -(1 H-1 ,2,4-triazol-1 -yl)-2-(2,4-difluorophenyl)- butan-2-ol
[R-(R*,R*)]-4-[2-[2-(2,4-Difluorophenyl)-2-hydroxy-1-methyl-3-(1H-1,2,4-triazol-1-yl)propyl]-4-thiazolyl]benzonitrile
4-[2-[(1R,2R)-2-(2,4-Difluorophenyl)-2-hydroxy-1-methyl-3-(1H-1,2,4-triazol-1-yl)propyl]-4-thiazolyl]benzonitrile
4-[2-[(1R,2R)-2-(2,4-Difluorophenyl)-2-hydroxy-1-methyl-3-(1H-1,2,4-triazol-1-yl)propyl]-4-thiazolyl]benzonitrile
Molecular Formula: C22H17F2N5OS
Molecular Weight: 437.47
Percent Composition: C 60.40%, H 3.92%, F 8.69%, N 16.01%, O 3.66%, S 7.33%
Eisai (INNOVATOR)
Properties: Colorless prisms from diisopropyl ether/n-hexane, mp 164-166°. [a]D24= -29.1° (c = 1.03 in methanol).
Melting point: mp 164-166°
Optical Rotation: [a]D24= -29.1° (c = 1.03 in methanol)
Therap-Cat: Antifungal.
http://www.google.com/patents/WO2011042827A1?cl=en…………m.p. 164-166° C; [a]=-30° (c=1 , methanol, 25° C); NMR (CDCI3): 1 .23(3H, d, J=8 Hz), 4.09(1 H, q, J=8Hz), 4.26(1 H, d, J=14Hz), 4.92(1 H, d, J=14Hz), 5.75(1 H, s), 6.75- 6.85(2H, m), 7.45-7.54(2H, m), 7.62(1 H, s), 7.69(1 H, s), 7.75(1 H, d, J=8Hz), 7.86(1 H, s), 8.03(1 H,d,J=8Hz). The analytical data were identical with published (US5648372 and Chem. Pharm. Bull. 1998, 46, 623-630).
Ravuconazole (BMS-207147 and ER-30346) is a potent triazole antifungal, being developed by Bristol-Myers Squibb, that is currently in phase I/II clinical trials.[1] The drug has a shown to have a similar spectrum of activity tovoriconazole, with an increased half-life.[2] However, ravuconazole has limited activity against species of Fusarium, Scedosporium, and Zygomycetes.[3][4]
 | |
| SYSTEMATIC (IUPAC) NAME | |
|---|---|
| 4-[2-[(2R,3R)-3-(2,4-Difluorophenyl)-3-hydroxy-4-(1,2,4-triazol-1-yl)butan-2-yl]-1,3-thiazol-4-yl]benzonitrile | |
| CLINICAL DATA | |
| LEGAL STATUS |
PHASE 2 AS ON SEPT 2014
|
| IDENTIFIERS | |
| CAS NUMBER | 182760-06-1 |
| ATC CODE | None |
| PUBCHEM | CID 467825 |
| NIAID CHEMDB | 057176 |
| CHEMICAL DATA | |
| FORMULA | C22H17F2N5OS |
| MOL. MASS | 437.465086 g/mol |
DRUG PROCESS…do not miss this
http://www.drugprocess.com/pdf/Isavuconazole_DPLA_ProcessSummary.pdf=++++++++++++++++++++++
…………………………………………………
Thiazole antifungals. III. Stereocontrolled synthesis of an optically active triazolymethyloxirane precursor to antifungal oxazolidine derivatives
Chem Pharm Bull 1991, 39(9): 2241
Chem Pharm Bull 1991, 39(9): 2241
……………………………………………………
Optically active antifungal azoles. I. Synthesis and antifungal activity of (2R,3R)-2-(2,4-difluorophenyl)-3-mercapto-1-(1H-1,2,4-triazol-1-yl)-2-butanol and its stereoisomers
Chem Pharm Bull 1993, 41(6): 1035
Chem Pharm Bull 1993, 41(6): 1035
………………………………………………………….
A novel route for chiral synthesis of the triazole antifungal ER-30346
Chem Pharm Bull 1998, 46(7): 1125
Chem Pharm Bull 1998, 46(7): 1125
……………………………………………………….
ER-30346 is synthesized by thiazole ring formation of (2R, 3R) -3- (2,4-difluorophenyl) -3-hydroxy-2-methyl-4- (1H-1,2,4-triazol-1-yl ) thiobutanamide (I) and 4-bromoacetylbenzonitrile (II) by means of reflux in methanol. The thioamide (I) is obtained with excellent yield from a chiral nitrile (III) by heating with diethyl dithiophosphate in aqueous medium.

Synthesis method
The nitrile (III), a chiral key intermediate of this synthesis, can be obtained by two different synthetic routes as follows: Route-a: The key step of this route is ring opening reaction of the trisubstituted oxirane (VII) by cyanide anion leading to the nitrile (III). The chiral oxirane (VII) is synthesized from (R) -lactic acid derivatives as already reported. The reaction of (VII) with diethylaluminum cyanide in toluene or lithium cyanide in tetrahydrofuran gives the nitrile (III) with high yield without any epimerization reaction.

The nitrile (III), a chiral key intermediate of this synthesis, can be obtained by two different synthetic routes as follows: Route-b: The starting material of this route is methyl (S) -3-hydroxy-2-methylpropionate (VIII ), which contains one additional carbon between the hydroxyl group and the 2-position carbon of (R) -lactate, the starting material of route-a. The hydroxyl group of (VIII) is protected by triphenylmethyl group. Then, 2,4 -difluorophenyl moiety is introduced to give the ketone (X). Direct conversion of the ketone (X) to the oxirane (XIV) by dimethylsulfoxonium methylide, the same condition for compound (IV) in route-a, does not proceed. The oxirane (XIV) having desired stereochemistry is obtained via oxidation reaction. The ketone (X) is converted to the exomethylene (XI) by Wittig reaction. The stereoselective oxidation of (XI) is achieved by means of osmium tetroxide in the presence of 4-methylmorpholine N-oxide to give the diol (XII) in 58% yield after separation of its epimer by column chromatography. After methanesulfonylation of the primary alcohol of (XII), a triazole moiety is introduced and the triphenylmethyl group is deprotected. Then, the primary hydroxyl group of (XVI) is oxidized under Swern oxidation condition to give the aldehyde (XVII), which is converted to the chiral nitrile intermediate (III) by means of heating with hydroxylamine-O-sulfonic acid.

The synthesis of (2S, 3S) -3- (2,4-difluorophenyl) -3-hydroxy-2-methyl-4- (1,2,4-triazol-1-yl) butyronitrile (XV), a key intermediate the synthesis of ER-30346 has been described: The tritylation of 3-hydroxy-2 (S) -methylpropionic acid methyl ester (I) with trityl chloride in hot pyridine gives the trityl ether (II), which is hydrolyzed with LiOH in H2O / THF / methanol yielding the free acid (III). The esterification of (III) with 2-mercaptopyridine (IV) by means of dicyclohexylcarbodiimide (DCC) in dichloromethane gives the thioester (V), which is treated with 2,4-difluorophenylmagnesium bromide (VI) in THF yielding the propiophenone (VII), which by treatment with methyltriphenylphosphonium bromide / NaH in THF is converted into the methylene derivative (VIII). The oxidation of (VIII) with OsO4 and N-methylmorpholine oxide in acetone affords, after column chromatography, the chiral diol (IX), which is monomesylated with mesyl chloride / triethylamine in dichlormethane giving the monoester (X). The reaction of (X) with 1,2,4-triazol (XI) and NaH in DMF yields (2R, 3S) -2- (2,4-difluorophenyl) -3-methyl-1- (1,2,4-triazol-1-yl) -4- (triphenylmethoxy) -2-butanol (XII), which is detritylated with p-toluenesulfonic acid in methanol affording the diol (XIII). The oxidation of (XIII) with oxalyl chloride / DMSO in dichloromethane gives the aldehyde (XIV), which is finally treated with hydroxylamine-O-sulfonic acid in water yielding the desired bytyronitrile intermediate (XV) already referenced.
Example 1
(2R,3R)-3-i4-(4-cvanophenyl)thiazol-2-yl1-1 -(1 H-1 ,2,4-triazol-1 -yl)-2-(2,4-difluorophenyl)- butan-2-ol
To a solution of racemic 3-[4-(4-cyanophenyl)thiazol-2-yl]-1 -(1 H-1 ,2,4-triazol-1 -yl)-2-(2,4- difluorophenyl)-butan-2-ol (43.7 g) in acetone (800 ml) a solution of (1 R)-10- camphorsulfonic acid (23 g) in methanol (300 ml) was added and the mixture was heated under reflux until a clear solution was obtained. The solution was slowly cooled to rt, seeded with crystals of the title enantiomeric salt and let overnight. The solid was collected by filtration, washed with acetone and dried to provide (2R,3R)-3-[4-(4- cyanophenyl)thiazol-2-yl]-1 -(1 H-1 ,2,4-triazol-1 -yl)-2-(2,4-difluorophenyl)-butan-2-ol (1 R)- 10-camphorsulfonate as white solid. This crude salt was then taken up in methylenechloride (100 ml) and water (ca. 100 ml) and the mixture was basified with aqueous sodium hydroxide solution. The organic layer was separated and the aqueous phase washed twice with methylenechloride (50 ml) and combined. The organic phases were then washed twice with water (2×50 ml), dried with sodium sulfate, filtrated and the solvent removed under reduced pressure. The crude product was then mixed with isopropanol (ca. 150 ml), heated for 10 min, cooled to 0° C and stirred for ca. 2 hrs. The product was collected, washed with isopropanol and dried under reduced pressure to provide the enantiomerically pure title compound (17.5 g, 41 % yield, 99.1 % ee); m.p. 164-166° C; [a]=-30° (c=1 , methanol, 25° C); NMR (CDCI3): 1 .23(3H, d, J=8 Hz), 4.09(1 H, q, J=8Hz), 4.26(1 H, d, J=14Hz), 4.92(1 H, d, J=14Hz), 5.75(1 H, s), 6.75- 6.85(2H, m), 7.45-7.54(2H, m), 7.62(1 H, s), 7.69(1 H, s), 7.75(1 H, d, J=8Hz), 7.86(1 H, s), 8.03(1 H,d,J=8Hz). The analytical data were identical with published (US5648372 and Chem. Pharm. Bull. 1998, 46, 623-630).
…………………………
Example 1
a) Preparation of (2R)-2′,5′-Difluoro-2-(3,4,5,6-tetrahydro-
2H-pyran-2-yloxy)-propiophenone A mixture of magnesium ( 7.25 g, 0.298 mol ) and iodine ( catalytic amount ) and l-bromo-2,5-difluorobenzene ( 20.0 g, 0.178 mol ) in THF ( 250ml ) was vigously stirred. The color of iodine was disappeared and the inner temperature rose up to 65°C. To this mixture was added additional l-bromo-2,5-difluorobenzene ( 30.0 g, 0.267 mol ) dropwise to maintain the inner temperature from 50 to 55°C over 45min. The resulting mixture was stirred at 55°C for 30min. then at r.t. for lhr. The – 21 -
mixture was cooled down to -5°C. To this mixture was added a solution of.4-[(2R)-2-(3,4,5,6-Tetrahydro-2H-pyran-2-yloxy)propionyl] morpholine ( 52.5 g, 0.216 mol ) in THF ( 150ml ) dropwise over 40min. And the resulting mixture was stirred at r.t. for 4hrs. The reaction mixture was cooled down to 5°C and saturated NH4C1 aq. ( 100ml ) was added carefully. The whole was diluted with H20 ( 600ml ) and extracted with EtOAc ( 400ml + 200ml x 2 ). The combined organic layer was dried over Na2S04 and concentrated in vacuo. The residue was chromatographed on silica gel ( n-hexane : EtOAc = 10 :1 ~ 5 : 1 ) to give (2R)-2′,5′- Difluoro-2-(3,4,5,6-tetrahydro-2H-pyran-2-yloxy)-propiophenone (47.3 g,
81 % ) as pale yellow syrup.
Physical form : colorless oil; FAB-MS: m/z 271(M+H)+; Η-NMR(CDCl;j): 1.42~1.90(9H,m),3.32~3.40(lHxl/2,m),3.69~3.77(lHxl/2,m),3.86~3.94 (lHxl/2,m),4.66(lHxl/2,t,J=3.6Hz),4.75(lHxl/2,t,J=3.6Hz),4.87(lHxl/2, q,J=6.6Hz),5.11(lHxl/2,q,J=6.9Hz),7.08~7.25(2H,m),7.49~7.55(lH,m).
b) Preparation of 2-(2,5-Difluorophenyl)-2-[(lR)-l-(3,4,5,6,- tetr ahy dro-2H-pyran-2-yloxy ) ethyl] oxir ane To a stirred mixture of NaH ( 60% in oil, 9.1g, 0.228mol ) in DMSO
(300ml ) was added portionwise trimethylsulfoxonium iodide ( 53.9g, 0.245 mol ) at the inner teperature with the range from 15°C to 18°C. over 20min. The ice bath was removed and the mixtuer was stirred at r.t. for 3hrs. The mixture was cooled down to 10°C. To this mixture was added a solution of (2R)-2′,5′-Difluoro-2-(3,4,5,6-tetrahydro-2H-pyran-2- yloxy)-propiophenone ( 47.3 g , 0.175 mol ) in DMSO (150ml ) dropwise over 20min. The resulting mixture was stirred at r.t. for 4hrs. The reaction mixture was poured into ice-water ( 800ml ). The whole was extracted with EtOAc ( 400ml + 200ml x 2 ). The combined organic layer was washed with brine, dried over Na2S04 and concentrated in vacuo.
The residue was chromatograkkphed on silicagel ( n-hexane : EtOAc = – 22 -
8 : 1 ~ 5 : 1 ) to give 2-(2,5-Difluorophenyl)-2-[(lR)-l-(3,4,5,6,- tetrahydro-2H-pyran-2-yloxy)ethyl]oxirane (48.3 g, 97 % ). Physical form : pale yellow syrup, EI-MS: m/z 284 (M)+ ; 1H-NMR(CDC13): 1.15(3Hxl/2,dd,J=6.6,1.3Hz), 1.24(3Hxl/2,dd, J=6.6,1.3Hz), 1.52-1.87 (6H,m),2.83~2,90(lH,m),3.07
(lHxl/2,d,J=5.3Hz),3.36(lHxl/2,d,J=5.6Hz), 3.48~3.56(lH,m),3.82~3.92 (lH,m),4.00~4.16(lH,m),4.73~4.92(lH,m), 6.96~7.02(lH,m),7.09~7.15 (lH,m).
c) Preparation of (3R)-2-(2,5-difluorophenyl)-3-(3,4,5,6- tetrahydro-2H-pyran-2-yloxy)-l-(lH-l,2,4-triazol-l-yl)-2-butanol
To a stirred suspension of NaH ( 60 % in oil, 21.0 g, 0.525 mol ) in DMF (300ml ) was added portionwise 1,2,4-triazole ( 43.3 g, 0.627 mol ) at the inner temperature from 2°C to 11°C over 30min. The resulting mixture was stirred at r.t. for l.δhrs. To this mixture was added a solution of 2-(2,5-Difluorophenyl)-2-[(lR)-l-(3,4,5,6-tetrahydro-2H- pyran-2-yloxy)ethyl]oxirane ( 48.3 g, 0.170 mol ) in DMF ( 50 ml ). The mixture was stirred at 60°C for lhr. and then at 65°C for 14hrs. The reaction mixture was cooled down to 10°C and then poured into ice- water (800 mL ). The resulting mixture was extracted with EtOAc
(400ml + 200ml x 2 ). The combined organic layer was dried over Na2S04 and concentrated in vacuo. The residue was chromatographed on silicagel ( n-hexane : EtOAc = 4 : 1 ~ 1 : 5 ) to give (3R)-2-(2,5- difluorophenyl)-3-(3,4,5,6-tetrahydro-2H-pyran-2-yloxy)-l-(lH-l,2,4- triazol-l-yl)-2-butanol ( 43.9 g, 73 % ) and recovered starting material
(13.2 g, 27 % ).
Physical form : colorless syrup ; FAB-MS: m/z 354 (M+H)+ ; Η- NMR(CDCl3): 1.00(3Hxl/2,d,J=6.6Hz),1.13(3Hxl/2,d,J=6.6Hz), 1.42~1.88(6H,m),3.38~3.60 (lH,m),3.80~4.00(lH,m),4.32~5.02(5H,m),6.83~6.99 (2H,m),7.14-7.21
(lH,m),7.73(lHxl/2,s),7.74(lHxl/2,s),7.92(lHxl/2,s),7.95(lHxl/2,s). – 23 -
d) Preparation of (2R,3R)-2-(2,5-difluorophenyl)-l-(lH-l,2,4- triazol-l-yl)-2,3-butanediol
A mixture of (3R)-2-(2,5-difluorophenyl)-3-(3,4,5,6-tetrahydro-2H- pyran-2-yloxy)-l-(lH-l,2,4-triazol-l-yl)-2-butanol ( 43.9 g, 0.124 mol ) and PPTS ( 15.6 g, 62.1 mmol ) in EtOH ( 400ml ) was stirred at 55°C for 5hrs. The mixture was was evaporated to remove solvent down to 100ml. The residue was poured into ice-aqueous NaHC03 ( 500ml ). The whole was extracted with EtOAc ( 400ml + 200ml x 2 ). The combined organic layer was dried over Na2S04 and concentrated in vacuo. The residue was chromatographed on silicagel (CH2C12 : MeOH = 20 : 1) to give (2R,3R)-2-(2,5-difluorophenyl)-l-(lH-l,2,4-triazol-l-yl)-2,3- butanediol (18.0 g, 54 % ). Physical form : colorless syrup ; FAB-MS: m/z 270 (M+H)’ ; ‘H- NMR(CDC13): 0.99(3H,d,J=6.6Hz),2.61(lH,d,J=10.6Hz), 4.31-4.36
(lH,m),4.79,4.88
(2H,ABq,J=14.5Hz),4.84(lH,s),6.84~6.99(2H,m),7.13~7.19(lH,m),7.84(l H,s),7.85(lH,s).
e) Preparation of (2R,3S)-2-(2,5-Difluorophenyl)-3-methyl-2-
[ ( 1H- 1 ,2,4-triazol-l -yl) -methyl] -oxir ane
To a cold ( 0°C ) and stirred solution of (2R,3R)-2-(2,5-difluorophenyl)- l-(lH-l,2,4-triazol-l-yl)-2,3-butanediol ( 35.0 g, 0.130 mol ) and triethylamine ( 54.8 ml, 0.393 mol ) in CH2C12 ( 500ml ) was added a mesylchloride ( 12.1 ml, 0.156 mol ) dropwise over 5min. The resulting mixture was stirred at r.t. for l.δhrs. The reaction mixture was poured into ice-water ( 300ml ). The resulting mixture was shaken well and the organic layer was separated. The aqueous layer was further extracted with CH2C12 ( 150ml x 2 ). All the organic layers were combined, dried over Na2SO4 and concentrated in vacuo to give mesylate ( 46.7 g ) as crude syrup. The obtained mesylate was dissolved in MeOH ( 500ml ) – 24 -
and the solution was cooled down to 0°C. To this solution was added 28% NaOMe methanol solution (29.0 ml ). The mixture was stirred at 0°C for 50min. The reaction mixture was evaporated to reduce the volume of the solvent down to 150 ml. The residue was poured into ice- water ( 300ml ). The resulting mixture was extracted with ethylacetate (300ml + 200ml x 2 ). The combined organic layer was dried over Na.,S0 and concentrated in vacuo. The residue was cromatographed on silicagel (hexane : EtOAc = 1 : 3 ) to give (2R,3S)-2-(2,5-Difluorophenyl)- 3-methyl-2-[(lH-l,2,4-triazol-l-yl)-methyl]-oxirane (30.3 g, 93 %).
Physical form : white solid ; FAB-MS : m z 252 (M+H)+ ; ]H- NMR(CDC13): 1.64(3H,d,J=5.6Hz),3.19(lH,q,J=5.6Hz),4.42,4.97 (2H,ABq,J=14.8Hz), 6.75~6.81(lH,m),6.89~7.01(2H,m),7.83(lH,s),7.98 UH,s).
f) Preparation of (2S,3R)-3-(2,5-Difluoro-phenyl)-3-hydroxy-
2-methyl-4-[l,2,4]triazol-l-yl-butyronitrile
A mixture of (2R,3S)-2-(2,5-Difluorophenyl)-3-methyl-2-[(lH-l,2,4- triazol-l-yl)-methyl]-oxirane ( 30.3 g, 0.121 mol ), trimethylsilylcyanide ( 65.0 ml ) and MgO ( 24.5 g ) in o-xylene ( 400 ml ) was stirred at 130°C for lOhrs. To this mixture was added additional trimethylsilylcyanide (20.0 ml ) and MgO ( 8.5 g ) and the resulting mixture was stirred at 130°C further for 6hrs. The reaction mixture was cooled down to r.t. The precipitate was filtered off and washed with CH2C12. The filtrate was concentrated in vacuo to give crude brown syrup.
This crude syrup was dissolved in THF ( 600ml ) and the solution was cooled down to 0°C. To this mixture was added 1.0 M tetra n- butylammoniumfluoride THF solution ( 133ml, 0.133 mol ) dropwise over 5min. The mixture was stirred at r.t. for 50min. The solvent was removed under reduced pressure down to 150ml. The residue was poured into ice-water ( 400ml ). The resulting mixture was extracted – 25 -
with EtOAc ( 300ml + 200ml x 2 ). The combined organic layer was dried over Na2SO4 and concentrated in vacuo. The residue was chromatographed on silicagel ( n-hexane : EtOAc = 1 : 3 ) to give (2S,3R)-3-(2,5-Difluoro-phenyl)-3-hydroxy-2-methyl-4-[l,2;4]triazol-l-yl- butyronitrile ( 30.5 g, 91 % ).
Physical form : colorless syrup ; FAB-MS : m/z 279 (M+H)+ ; Η- NMR(CDCl3): 1.19(3H,d,J=7.3Hz),3.33(lH,q,J=7.3Hz),4.82,5.00 (2H,ABq,J=13.9Hz), 5.56(lH,brs),6.89~7.04(2H,m),7.12~7.19(lH,m),7.85(lH,s),7.86(lH,s).
g) Preparation of (2R,3R)-3-(2,5-Difluoro-phenyl)-3-hydroxy-
2-methyl-4- [ 1 ,2,4] triazol-1 -ylthiob tyramide
A mixture of (2S,3R)-3-(2,5-Difluoro-phenyl)-3-hydroxy-2-methyl-4- [l,2,4]triazol-l-yl-butyronitrile ( 30-5 S> O.llOmol ), diethyldithio- phospate ( 235 ml ) and H2O ( 110 ml ) was stirre at 80°C for 2hrs. The reaction mixture was cooled down to r.t. n-Hexane ( 400ml ) and water (200 ml ) was added. The whole was shaken well and the aqueous layer was separated. The remaining organic layer was further extracted with H20 ( 100ml x 3 ). All the aqueous layer was combined. Cooled down to
0°C and neutralized and basified ( PH8 ) with NaHC03. This basic(PH8) aqueous layer was extracted with EtOAc ( 300ml + 100ml x 3 ). The combined organic layer was dried over Na2S04 and concentrated in vacuo to give dark brown syrup. By addition of CH2C12 ( 100ml ) to this crude syrup, precipitate was formed. The precipitate was filtered and washed with CH2C12-hexane ( 5 : 1 mixture ) to give (2R,3R)-3-(2,5-Difluoro-phenyl)-3-hydroxy-2-methyl-4-[l,2,4]triazol-l- ylthiobutyramide ( 19.2 g, 56 % ) as white powder. On the oter hand, the filtrate was concentrated in vacuo and the residue was chromatographed on silica gel ( Wako-gel C-300, CH2C12 : MeOH = 20 :
1 ) to give additional (2R,3R)-3-(2,5-Difluoro-phenyl)-3-hydroxy-2- – 26 -
methyl-4-[l,2,4]triazol-l-ylthiobutyramide ( 7.46 g, 22 % ) as pale brown amorphous powder.
Physical form : White solid ; FAB-MS : m/z 313 (M+H)+ ; ‘H-NMR (CDC13): 1.12(3H,d,J=7.3Hz),3.74(lH,q,J=7.3Hz), 4.55,5.12 (2H,ABq,J=14.5Hz), 5.84(lH,s),6.85~7.02(2H,m),7.15-7.22(lH,m),7.80
(1H,S),7.89(1H,S), 7.89(lH,brs),8.43(lH,brs).
h) Preparation of 4-{2-[(lR,2R)-2-(2,5-Difluoro-phenyl)-2- hydroxy-l-methyl-3-[l,2,4]triazol-l-yl-propyl]-thiazol-4-yl}- benzonitrile
A mixture of (2R,3R)-3-(2,5-Difluoro-phenyl)-3-hydroxy-2-methyl-4- [l,2,4]triazol-l-ylthiobutyramide ( 26.7 g, 85.4 mmol ) and a-bromo-4′- cyano-acetophenone ( 24.0 g, 0.107 mol ) in EtOH ( 500ml ) was refluxed for lhr. The reaction mixture was cooled down to r.t. And the solvent was removed under reduced pressure down to 150ml. The residue was poured into in to cold ( 0°C ) saturated NaHC03 aq. ( 400ml ). The resulting mixture was extracted with EtOAc ( 300ml + 150 ml x 2 ). The combined organic layer was washed with brine (200ml ), dried over Na2S04 and concentrated in vacuo. The residue was chromatographed on silica gel ( Wako-gel C-300, Hexane : EtOAc = 1 : 2 ) to give 4-{2-
[(lR,2R)-2-(2,5-Difluoro-phenyl)-2-hydroxy-l-methyl-3-[l,2,4]triazol-l- yl-propyl]-thiazol-4-yl}-benzonitrile ( 32.0 g, 86 % ).
Physical form : colorless heavy syrup ; ESI-MS : m/z 437 (M)+ ; ‘H-
NMR(CDCl3): 1.25(3H,d,J=7.3Hz),4.12(lH,q,J=7.3Hz),4.26,4.96 (2H,Abq,J=14.5Hz), 5.75(lH,s),6.89~7.07(2H,m),7.23~7.29(lH,m),7.65
(lH,s),7.71(lH,s),7.75,8.02 (4H,Abq,J=8.6Hz),7.85(lH,s). – 27 -
i) Preparation of 4-{4-[(tert-Butoxycarbonyl-methyl-amino)- acetoxy]-3,5-dimethyl-benzyl}-l-[(2R,3R)-3-[4-(4-cyano-phenyl)- thiazol-2-yl]-2-(2,5-difluoro-phenyl)-2-hydroxy-butyl]-lH- [l,2,4]triazol-4-ium bromide A mixture of 22.7mg of 4-{2-[(lR,2R)-2-(2,5-Difluoro-phenyl)-2-hydroxy- l-methyl-3-[l,2,4]triazol-l-yl-propyl]-thiazol-4-yl}-benzonitrile and 25.0mg of 4-tert-butoxycarbonyl-methyl-aminoacetoxy-3,5-dimethyl- benzyl bromide in CH3CN(1.5mL) was refluxed over 15hrs. The solvent was evaporated in vacuo and the residue was chromatographed on silica gel (Wakogel C-200, solvent:CH2Cl MeOH=10/l) to give 4-{4-[(tert-
Butoxycarbonyl-methyl-amino)-acetoxy]-3,5-dimethyl-benzyl}-l- [(2R,3R)-3-[4-(4-cyano-phenyl)-thiazol-2-yl]-2-(2,5-difluoro-phenyl)-2- hydroxy-butyl]-lH-[l,2,4]triazol-4-ium bromide (36.0mg, 84% as colorless heavy syrup) ; FAB-MS : m/z 743 (M-Br)’ ; Η-NMR(CDC1S): 1.23(3H,d,J=7.3Hz),
1.47(9H,s),2.14(6H,s),3.03(3H,s),4.15(lH,q,J=7.3Hz),4.25(2H,s), 4.98,5.16(2H,ABq,J=13.9Hz),5.39~5.54(2H,m),6.27(lH,s),6.89-7.07(4H, m),7.24~7.27(lH,m),7.58(lH,s),7.73,8.06(4H,ABq,J=8.58),8.07(lH,s),ll. 26 (lH,s).
j) Preparation of l-{(2R,3R)-3-[4-(4-cyano-phenyl)-thiazol-2-yl]- 2-(2,5-difluoro-phenyl)-2-hydroxy-butyl}-4-(3,5-dimethyl-4- methylaminoacetoxy-benzyl)-lH-[l,2,4]triazol-4-ium bromide To a solution of 36mg of 4-{4-[(tert-Butoxycarbonyl-methyl-amino)- acetoxy]-3,5-dimethyl-benzyl}-l-[(2R,3R)-3-[4-(4-cyano-phenyl)-thiazol-
2-yl]-2-(2,5-difluoro-phenyl)-2-hydroxy-butyl]-lH-[l,2,4]triazol-4-ium bromide in ethylacetate(2ml) was added dropwise 4N HC1 ethylacetate solution(lmL) and the mixture was stirred at r.t. for 4hrs.The precipitate was “filtered and washed with diethylether to give 1- {(2R,3R)-3-[4-(4-cyano-phenyl)-thiazol-2-yl]-2-(2,5-difluoro-phenyl)-2- hydroxy-butyl}-4-(3,5-dimethyl-4-methylaminoacetoxy-benzyl)-lH- – 28 -
[l,2,4]triazol-4-ium bromide (24.5mg, 74% as HC1 salt and as white solid) ;
FAB-MS : m/z 643 (M-Br)+ ; Η-NMR(DMSO-d): 1.19(3H,d,J=7.3Hz), 2.11(6H,s),2.64(3H,s),4.15(lH,q,J=7.3Hz),4.41(2H,s),4.74,5.04(2H,ABq,J =14.5Hz),5.40(2H,s),6.76(lH,brs),7.10(2H,s),7.20~7.38(2H,m), 7.94,8.21
(4H,ABq,J=8.25),8.45(lH,s),9.07(lH,s),9.50(lH,brs),10.17(lH,s).
………………………
OR
COMPD 21
- Example 88:Preparation of a compound of the structural formula:
-
-
2-(2,4-Difluorophenyl)-3-thioamide-1-(1H-1,2,4-triazol-1-yl)-2-butanol (the raw material 2) (156 mg) was dissolved in EtOH (2 ml), and 2-bromo-4′-cyanoacetophenone (the raw material 3) (224 mg) was added to the solution, followed by heating and refluxing for 1 hour. The liquid reaction mixture was neutralized with a saturated aqueous solution of NaHCO3 and subjected to extraction with AcOEt. After the extract was washed with H2O and then a saturated aqueous solution of NaCl and dried over MgSO4, AcOEt was distilled out. The resultant residue was purified by chromatography on silica gel (SiO2: 20 g, eluted with CH2Cl2 and then with 1% solution of MeOH in CH2Cl2), and then crystallized from IPE, thereby obtaining the intended compound (109 mg). Physical properties of this compound are described below.
- mp:
- 196-197°C.
- NMR:
- δ solvent (CDCl3)
1.23(3H,d,J=8.0Hz), 4.09(1H,q,J=8.0Hz), 4.26(1H,d,J=14.3Hz), 4.92(1H,d,J=14.3Hz), 5.74(1H,s), 6.78-6.85(2H,m), 7.48-7.54(1H,m), 7.64(1H,s), 7.69(1H,s), 7.75(1H,d,J=8.1Hz), 7.85(1H,s), 8.03(1H,d,J=8.1Hz). - MS:
- MH+ = 438.

References
- National Cancer Institute. Ravuconazole in Preventing Fungal Infections in Patients Undergoing Allogeneic Stem Cell Transplantation. In: ClinicalTrials.gov [Internet]. Bethesda (MD): National Library of Medicine (US). 2000- [cited 2010 Feb 18]. Available from:http://clinicaltrials.gov/ct2/show/NCT00064311?term=ravuconazole&spons_ex=Y&rank=1 NLM Identifier: NCT00064311.
- The Aspergillus Website, Pasqualotto AC, Denning DW. Ravuconazole. Date accessed: 2010 Feb 18.
- Pasqualotto AC, Thiele KO, Goldani LZ (2010). “Novel triazole antifungal drugs: focus on isavuconazole, ravuconazole and albaconazole”. Curr Opin Investig Drugs 11 (2): 165–74. PMID 20112166.
- Pfaller, M. A.; Messer, S. A.; Hollis, R. J.; Jones, R. N.; Sentry Participants, Group (2002). “Antifungal Activities of Posaconazole, Ravuconazole, and Voriconazole Compared to Those of Itraconazole and Amphotericin B against 239 Clinical Isolates of Aspergillus spp. and Other Filamentous Fungi: Report from SENTRY Antimicrobial Surveillance Program, 2000″. Antimicrobial Agents and Chemotherapy46 (4): 1032.doi:10.1128/AAC.46.4.1032-1037.2002. PMC 127116. PMID 11897586.
Literature References:
Ergosterol biosynthesis inhibitor. Prepn (stereochemistry unspecified): T. Naitoet al, EP 667346; eidem,US 5648372 (1995, 1997 both to Eisai); of optically acitve form: A. Tsuruoka et al., Chem. Pharm. Bull. 46, 623 (1998). Chiral synthesis: Y. Kaku et al., ibid. 1125.
In vitro comparative antifungal spectrum: J. C. Fung-Tomc et al., Antimicrob. Agents Chemother. 42, 313 1998. Antifungal activity in candidosis: K. V. Clemons, D. A. Stevens, ibid. 45, 3433 (2001); in aspergillosis: W. R. Kirkpatrick et al., J. Antimicrob. Chemother. 49, 353 (2002).
Clinical evaluation in onychomycosis: A. K. Gupta et al., J. Eur. Acad. Dermatol. Venereol. 19, 437 (2005).
Review of development and therapeutic potential: S. Arikan, J. H. Rex, Curr. Opin. Invest. Drugs 3, 555-561 (2002).
Extras you may need
Scheme 1 :

The manufacturing process for Isavuconazole is similar: Since Isavuconazole differentiates from Ravuconazole by only another fluorine substitution on the aromatic ring (2,5- instead of 2,4-difluorophenyl), the identical synthesis has been used (US 6300353 from October 9, 2001 and Bioorg. & Med. Chem. Lett. 13, 191 (2003)). Consequently, also this manufacturing process, based on (R)-lactic acid, faces the same problems: to many steps, extremely low overall yield and in addition to US patent 6300353 claims even already known step as novel (claim 36).
Recent attempts to improve this concept as reported in WO 2007/062542 (Dec.1 , 2005), using less expensive, natural configured (S)-lactic acid, also failed: As already reported in US 6133485 and in US 2003/0236419, the second chiral center was formed from an optically active allyl alcohol prepared in a few steps from (S)-lactic acid. This allyl alcohol was subjected to Sharpless diastereoselective epoxidation providing first an opposite configured, epimeric epoxy alcohol which had to be then epimerized in an additional inversion step yielding finally the desired epoxy alcohol as the known precursor for Isavuconazole (US 6300353). It is obvious that this process using less expensive (S)- lactic acid makes the entire process with an inversion step even more complex than the original approach.
Elegant and more efficient process has been claimed in US 2004/0176432 from June 26, 2001 ) in which both chiral centers have been formed simultaneously, diastereo- and enantio-selectively pure in one single reaction step using chiral (R)-2-butynol as a chiral precursor in the presence of Pd(ll)-catalyst and diethyl zinc (Scheme 2).
Scheme 2:

Since water soluble, (R)-2-butynol is expensive, recently identical process has been published, in which instead of (R)-2-butynol less water soluble and therefore, less expensive (R)-4-phenyl-3-butyn-2-ol was used (Synthetic Commun. 39, 161 1 (2009)). Nevertheless, as incorrectly stated there, this process does not provide better diastereoselectivity than the original process using (R)-2-butynol: On the contrary disadvantage of this process is a very bad atom economy because huge phenyl group of (R)-4-phenyl-3-butyn-2-ol has to be “disposed” in oxidation step by the conversion of triple bond into carboxylic acid function.
……………………………
The invention relates to a process for the manufacture of a
diastereomerically and enantiomerically enriched ester intermediate for isavuconazole or ravuconazole.
Isavuconazole and ravuconazole are triazole antifungal compounds. Processes for the manufacture of isavuconazole and ravuconazole were disclosed in patents WO99/45008, WO2007/062542 and WO03/002498 to Basilea. In WO2011/042827 a process for the manufacture of enantiomerically pure antifungal azoles such as ravuconazole and isavuconazole is disclosed, wherein a classical resolution of a racemic mixture is performed by the addition of an enantiopure chiral acid, then collection of the desired diastereomer followed by conversion of the salt into the enantiomerically pure form of the desired compound by treatment with a base or an ion-exchange resin. The disadvantages of using such classical resolution are that the chiral auxiliary needs to be applied in near stoichiometric amounts, and that additional process steps are required for recovery of these relatively high amounts of chiral reagent as well as for converting the salt into the free enantiopure product.
Reaction Scheme 1:

7
EFINACONAZOLE(JUBLIA)
Efinaconazole
(2R,3R)-2-(2,4-Difluorophenyl)-3-(4-methylene-1-piperidinyl)-1-(1H-1,2,4-triazol-1-yl)-2-butanol
(2R, 3R) -2 – (2,4 – difluorophenyl) -3 – (4 – methylene-piperidin-1 – yl) -1 – (1H-1, 2,4 – triazol-1 – yl) butan-2 – manufacture ol (KP-103)
Efinaconazole is a triazole antifungal. It is approved for use in Canada as 10% topical solution for the treatment of onychomycosis(fungal infection of the nail).[1][2] Efinaconazole acts as a 14α-demethylase inhibitor.[3]
| IDENTIFIERS | |
|---|---|
| CAS NUMBER | 164650-44-6 |
| PUBCHEM | CID 489181 |
| CHEMSPIDER | 428538 |
| CHEMICAL DATA | |
| FORMULA | C18H22F2N4O |
| MOL. MASS | 348.39 g/mol |
SEE AT
LAVAL, Quebec, June 9, 2014 /PRNewswire/ — Valeant Pharmaceuticals International, Inc. (NYSE: VRX) (TSX: VRX) today announced that that its wholly owned subsidiary, Valeant Pharmaceuticals North America LLC, received notice that the U.S. Food and Drug Administration (FDA) has approved the New Drug Application (NDA) for Jublia® (efinaconazole 10% topical solution), the first topical triazole approved for the treatment of onychomycosis of the toenails
EFINACONAZOLE,
KP-103, 164650-44-6, Efinaconazole [INN], UNII-J82SB7FXWB, SureCN300738, AC1LAJ21, Efinaconazole [USAN:INN], CHEMBL2103877
(2R,3R)-2-(2,4-difluorophenyl)-3-(4-methylidenepiperidin-1-yl)-1-(1,2,4-triazol-1-yl)butan-2-ol
Molecular Formula: C18H22F2N4O Molecular Weight: 348.390286
……………………………….
PATENT
Method for producing butanol derivatives – 1 – 2 – triazole Is a (compound described in Example 1 of Patent Document 1) a compound of formula 1 to be effective against fungal diseases of humans and animals are known, the present invention, (2R, 3R) – 2 – (2,4 – difluorophenyl) -3 – (4 – methylene piperidin-1 – yl) -1 – (1H-1, 2,4 – triazol-1 – yl) butan-2 – (generic name ol ( The present invention relates to preparation of their salts that Fina et Kona zole (Efinaconazole)), hereinafter abbreviated as “KP-103″ or even) in: INN).


Production Example 1
Methanol / water mixture methylene piperidine (4-MP) – 4 obtained by the method described in the manufacture WO 97/11939 pamphlet methylene piperidine hydrobromide salt of (4-MP · HBr) – 4 was cooled by stirring in an ice bath under a solution 0.8M 500mL (0.4mol). Thereafter, the solution is added in several portions (0.36mol) 48% hydrobromic acid 61.3g, followed by stirring for 1 hour in an ice bath. Thereafter, to precipitate white crystals by evaporating the solvent by heating under reduced pressure.Subsequently, we conducted two times operation for azeotropic water by distilling off the solvent by heating under reduced pressure and toluene was added to 50mL, and added acetone 192mL, was 2 hours under stirring ice bath. Thereafter, The crystals are filtered, washed crystals with 60mL (cooled in an ice bath) of acetone, 4-MP · HBr58g as colorless crystals (yield: 90%) After air-drying at room temperature, for 12 hours and dried under reduced pressure at 40 ℃ I got.
1 H-NMR (500MHz, CDCl 3)
δ: 2.62 (4H, t, J = 6.09 Hz), 3.26 (4H, t, J = 6.09 Hz), 4.90 (2H, s), 9.18 (1H, br).
Melting point (DSC): 147 ~ 147.9 ℃


The method for obtaining the amino alcohol by ring-opening addition reaction of the amine to the epoxide, in general, using a large excess of amine, and is performed for a long time at a high temperature. In the conventional method, in order to use the amine of the large excess of byproducts is large and requires a recovery step of an amine, also in terms of production costs, it is desirable as a production method on an industrial scale if the amine is expensive no. In order to increase the reactivity of the reaction, the reaction using a Lewis acid have been proposed, also, difficult to use industrially Lewis acid to be used is unstable or expensive, perchlorate, etc., are those toxic-risk is less secure high, there is a problem such as needing attention in use (Non-Patent Documents 1 and 2). It is also reported that could be the use of lithium bromide, to enhance the reactivity under solvent-free conditions at room temperature (Non-Patent Document 3). It is believed that since the liquid at normal temperature and epoxides, amines are used, the method reported in the literature, was achieved by reaction at a high concentration under solvent-free conditions starting material. Thus, a solid at room temperature and can not be applied to epoxides and amines, particularly high melting point.
On the other hand, as described in Patent Document 1, formula 1 compound is produced by ring-opening addition reaction of the amine to the epoxide. In this production process, as the epoxide (2R, 3S) -2 – (2,4 – difluorophenyl) -3 – methyl -2 – [(1H-1, 2,4 - triazol-1 - yl) methyl] oxirane and ( used that methylene piperidine (hereinafter, abbreviated as “4-MP” also) some – is used will hereinafter be abbreviated as “epoxy triazole” also) some, 4 as the amine. In this manufacturing process, it has the disadvantage for heating under reflux for a long time by using the 4-MP solution in large excess in the ring-opening addition reaction, and it is necessary to by-products are produced much in the reaction step to remove them. Furthermore, 4 – methylene-piperidine is prepared by the method described in Patent Document 2, but the purity is low because it is obtained in an aqueous solution, and also affects the reactivity when the distillation isolation there is a problem of impurities by heat generated.
WO 94/26734 pamphlet WO 97/11939 pamphlet
Synthesis, 2004, No.10, pp 1563-1565 J. Org. Chem., 2007, vol. 72, pp 3713-3722 Eur. J. Org. Chem., 2004, No.17, pp 3597-3600
The purpose of the present invention, (2R, 3S) -2 – to oxirane (2,4 – difluorophenyl) -3 – methyl-2 – [methyl-(yl 1H-1, 2,4 - - -1 triazole)] that without using methylene piperidine may yield a compound of Formula 1 under mild conditions to provide a manufacturing method with a reduced formation of by-product – 4 large excess of ring-opening addition reaction of methylene piperidine – 4 some.
As a result of intensive investigations, 4 the present inventors found that – if the acid addition salt of methylene-piperidine, 4 – impurities incorporated in the acquisition phase of the methylene piperidine has been removed, will be isolated as a solid high purity It can, therefore, four of the starting raw material in the ring-opening addition reaction of the amine to epoxy triazole – and that is able to increase the purity of methylene piperidine, the reaction solvent, the ring-opening addition reaction of amines to the epoxy triazole medium, is performed in the presence of hydroxides of alkali metals or alkaline earth metals in particular, 4 – there is no need to use excess methylene piperidine, high yield, by-products and a compound of Formula 1 under mild conditions to discover that it can be produced by reducing things, and have completed the present invention.
I will explain in detail the methods of the present invention are described below.
As indicated by the following reaction formula, the present invention, (2R, 3S) -2 – (2,4 – difluorophenyl) -3 – methyl -2 – [(1H-1, 2,4 - triazol - hydrate thereof or a hydroxide of an alkali metal or alkaline earth metal is selected from the group the reaction solvent, consisting of strontium lithium, sodium, and calcium, and a methylene piperidine acid addition salt and 4 - yl) methyl] oxirane including the presence of a, is reacted relates to a manufacturing method of the formula (1) compound.
As indicated by the following reaction formula, the present invention, (2R, 3S) -2 – (2,4 – difluorophenyl) -3 – methyl -2 – [(1H-1, 2,4 - triazol - hydrate thereof or a hydroxide of an alkali metal or alkaline earth metal is selected from the group the reaction solvent, consisting of strontium lithium, sodium, and calcium, and a methylene piperidine acid addition salt and 4 - yl) methyl] oxirane including the presence of a, is reacted relates to a manufacturing method of the formula (1) compound.

(Wherein HX represents an acid addition salt of the acid)
Starting material of the process of the present invention
It can be also performed by using a starting compound of any amount ranging ton level from the g level, the method of the present invention may be determined the amount of solvent depending on the amount of the starting compound used.
It can be also performed by using a starting compound of any amount ranging ton level from the g level, the method of the present invention may be determined the amount of solvent depending on the amount of the starting compound used.
(2R, 3S) -2 – (2,4 – difluorophenyl) -3 – methyl -2 – [(1H-1, 2,4 - -1 triazole - yl) methyl] oxirane, JP 2-191262 issue It can be obtained by methods described in the Gazette.
4 – is represented by the following formula methylenepiperidine acid addition salts:
4 – is represented by the following formula methylenepiperidine acid addition salts:

Wherein an acid of the acid addition salts with HX, 4 – The acid forming the methylene piperidine acid addition salts may, for example, hydrochloric, hydrobromic, if an acid that forms a salt with an amine is basically inorganic acid acid, hydroiodic acid, sulfuric acid, nitric acid, phosphoric acid, boric acid, chloric acid, carbonic acid, etc.; formic acid, acetic acid, trifluoroacetic acid, propionic acid, oxalic acid, methanesulfonic acid, benzenesulfonic acid, p – organic acids such as toluenesulfonic acid and the like, but is not limited thereto. Hydrochloric acid, hydrobromic acid, hydroiodic acid, nitric acid, trifluoroacetic acid and the like, more preferably, preferred examples of the acid is a hydroiodic acid or hydrobromic acid.
4 – methylene-piperidine acid addition salt, 4 – can be obtained by reacting a conventional manner with an acid corresponding to the acid addition salt with methylene piperidine.
Here, the 4 – methylenepiperidine may be preferable in terms of production on an industrial scale, prepared by the method described in WO 97/11939 pamphlet. 4 is manufactured here – methylene-piperidine, and also are obtained in the form of an aqueous solution, impurities produced by heat during the distillation isolation is included, according to the manufacturing method described below, 4 – methylene-piperidine the impurities are removed, acid addition salts can be isolated as a solid high purity.
That is, 4 – preferred method of methylenepiperidine acid addition salts, the following steps:
(1) 4 – reacting with an acid corresponding to the acid addition salt, a solution of methylene-piperidine, and (2) the solvent is evaporated as necessary, washed suspension crystallization or a product obtained , it is a method comprising the step of purifying.
(1) 4 – reacting with an acid corresponding to the acid addition salt, a solution of methylene-piperidine, and (2) the solvent is evaporated as necessary, washed suspension crystallization or a product obtained , it is a method comprising the step of purifying.
Here, 4 (1) Step – A solution of methylene piperidine solution in a mixed solvent of alcohol or water and aqueous alcohol solution or (such as methanol), and the like. 0.9-1.0 equivalents is preferably used amount of the methylene piperidine – 4 of the acid corresponding to the acid addition salt. Reaction conditions (1) is carried out at room temperature from 0 ℃, the reaction time is several hours 15 minutes.
After the step (1), if necessary, by conventional methods, for example, under reduced pressure, the temperature is carried out by heating from room temperature solvent was evaporated. In the case of decreasing the water content of the reaction system, for example, by azeotropic toluene or use of the desiccant.
How to purify washed suspension or crystallization in step (2) The method of cleaning is suspended in a solvent crystals or recrystallized after being dissolved in a solvent, obtained by filtration, or distilling off the solvent I may be mentioned.
After the step (1), if necessary, by conventional methods, for example, under reduced pressure, the temperature is carried out by heating from room temperature solvent was evaporated. In the case of decreasing the water content of the reaction system, for example, by azeotropic toluene or use of the desiccant.
How to purify washed suspension or crystallization in step (2) The method of cleaning is suspended in a solvent crystals or recrystallized after being dissolved in a solvent, obtained by filtration, or distilling off the solvent I may be mentioned.
The acid addition salts, conditions of the production method is different, for example, after the reaction of step (1), the solvent was evaporated, the case of hydrochloride and hydrobromide and acetone crystals was then obtained After washing the suspension and filtered. For p-toluenesulfonate, After the reaction of step (1), the solvent was evaporated, and dissolved in ethyl acetate (10:1) / isopropanol mixture and the residue is recrystallized. For nitrate hydroiodide, and trifluoroacetic acid salt, after the reaction of step (1) to dryness by distilling off the solvent, washed and suspended by addition of diisopropyl ether to the residue.
Reaction conditions of the process of the present invention
4 – triazole for the epoxy, the amount of methylene piperidine acid addition salt is 1 to 5 equivalents, preferably 1 to 1.5 equivalents.
4 – triazole for the epoxy, the amount of methylene piperidine acid addition salt is 1 to 5 equivalents, preferably 1 to 1.5 equivalents.
As the hydroxide of alkali metal or alkaline earth metal in the reaction of the present invention, a hydrate thereof or strontium hydroxide lithium hydroxide, sodium hydroxide, calcium hydroxide and the like. More preferably, lithium hydroxide, a hydrate thereof or calcium hydroxide, more preferably a hydrate thereof or a lithium hydroxide.
The amount of the hydroxide of alkaline earth metals varies depending on the basicity and the type of compound used or the alkali metals, 4 – for methylenepiperidine acid addition salt is 1 to 5 equivalents usually preferably is 1 to 1.5 equivalents.
Production Example 1
Methanol / water mixture methylene piperidine (4-MP) – 4 obtained by the method described in the manufacture WO 97/11939 pamphlet methylene piperidine hydrobromide salt of (4-MP · HBr) – 4 was cooled by stirring in an ice bath under a solution 0.8M 500mL (0.4mol). Thereafter, the solution is added in several portions (0.36mol) 48% hydrobromic acid 61.3g, followed by stirring for 1 hour in an ice bath. Thereafter, to precipitate white crystals by evaporating the solvent by heating under reduced pressure.Subsequently, we conducted two times operation for azeotropic water by distilling off the solvent by heating under reduced pressure and toluene was added to 50mL, and added acetone 192mL, was 2 hours under stirring ice bath. Thereafter, The crystals are filtered, washed crystals with 60mL (cooled in an ice bath) of acetone, 4-MP · HBr58g as colorless crystals (yield: 90%) After air-drying at room temperature, for 12 hours and dried under reduced pressure at 40 ℃ I got.
1 H-NMR (500MHz, CDCl 3)
δ: 2.62 (4H, t, J = 6.09 Hz), 3.26 (4H, t, J = 6.09 Hz), 4.90 (2H, s), 9.18 (1H, br).
Melting point (DSC): 147 ~ 147.9 ℃
Production Example 2
Water removal operation (4-MP) methylene piperidine – 4 obtained by the method described in the manufacture WO 97/11939 pamphlet methylene piperidine p-toluenesulfonic acid salt of (4-MP · PTSA) – 4 isopropanol (9.7g, 0.1mol) of (IPA) in (50mL) solution, 4-MP, which is subjected to, the resulting p-toluenesulfonic acid monohydrate (PTSA · H 2 O) (18.1g, 0.095mol) / was added (80mL) IPA, (weak exothermic) after which the mixture was stirred for 30 minutes at room temperature, evaporated under reduced pressure IPA, and was heated and dissolved in (250mL) with ethyl acetate / IPA mixture = 10:1 residue. After cooling to room temperature and allowed to stand for 20 hours at 0 ~ 5 ℃, filtered washing the precipitated crystals were obtained (91.2% yield) 4-MP · PTSA 23.34g of white crystals to dry.
1 H-NMR (400MHz, DMSO-d 6)
δ: 2.29 (3H, s), 2.35 (4H, t, J = 6.4 Hz), 3.08 (4H, t, J = 6.4 Hz), 4.85 (2H, s), 7.13 (2H, d, J = 8.2 Hz ), 7.49 (2H, d, J = 8.2 Hz), 8.58 (2H, brs).
Water removal operation (4-MP) methylene piperidine – 4 obtained by the method described in the manufacture WO 97/11939 pamphlet methylene piperidine p-toluenesulfonic acid salt of (4-MP · PTSA) – 4 isopropanol (9.7g, 0.1mol) of (IPA) in (50mL) solution, 4-MP, which is subjected to, the resulting p-toluenesulfonic acid monohydrate (PTSA · H 2 O) (18.1g, 0.095mol) / was added (80mL) IPA, (weak exothermic) after which the mixture was stirred for 30 minutes at room temperature, evaporated under reduced pressure IPA, and was heated and dissolved in (250mL) with ethyl acetate / IPA mixture = 10:1 residue. After cooling to room temperature and allowed to stand for 20 hours at 0 ~ 5 ℃, filtered washing the precipitated crystals were obtained (91.2% yield) 4-MP · PTSA 23.34g of white crystals to dry.
1 H-NMR (400MHz, DMSO-d 6)
δ: 2.29 (3H, s), 2.35 (4H, t, J = 6.4 Hz), 3.08 (4H, t, J = 6.4 Hz), 4.85 (2H, s), 7.13 (2H, d, J = 8.2 Hz ), 7.49 (2H, d, J = 8.2 Hz), 8.58 (2H, brs).
Production Example 3
4 obtained by the method described in the manufacture WO 97/11939 pamphlet methylene piperidine hydrochloride (4-MP · HCl) – – 4 subjected to moisture removal operation methylene piperidine (4-MP), obtained was cooled by stirring in an ice bath under (4.12mol) 4-MP 400g that is. Thereafter, the solution was added concentrated hydrochloric acid and 350mL (4.08mmol), and the mixture was stirred in an ice bath. After concentration under reduced pressure was performed 3 times operation for azeotropic water and concentrated under reduced pressure and toluene was added to 300mL. The washed suspension under ice-cooling and addition of acetone 300mL. The filtered crystals were washed with acetone crystals, 4 and dried under reduced pressure at room temperature – was obtained (46% yield) methylene-piperidine hydrochloride (4-MP · HCl) 336.8g.
1 H-NMR (500MHz, CDCl 3)
δ: 2.58 (4H, t, J = 6.1Hz), 3.22 (4H, t, J = 6.1Hz), 4.89 (2H, s), 9.70 (1H, br s).
4 obtained by the method described in the manufacture WO 97/11939 pamphlet methylene piperidine hydrochloride (4-MP · HCl) – – 4 subjected to moisture removal operation methylene piperidine (4-MP), obtained was cooled by stirring in an ice bath under (4.12mol) 4-MP 400g that is. Thereafter, the solution was added concentrated hydrochloric acid and 350mL (4.08mmol), and the mixture was stirred in an ice bath. After concentration under reduced pressure was performed 3 times operation for azeotropic water and concentrated under reduced pressure and toluene was added to 300mL. The washed suspension under ice-cooling and addition of acetone 300mL. The filtered crystals were washed with acetone crystals, 4 and dried under reduced pressure at room temperature – was obtained (46% yield) methylene-piperidine hydrochloride (4-MP · HCl) 336.8g.
1 H-NMR (500MHz, CDCl 3)
δ: 2.58 (4H, t, J = 6.1Hz), 3.22 (4H, t, J = 6.1Hz), 4.89 (2H, s), 9.70 (1H, br s).
Preparation Example 4
Methanol / water mixture methylene piperidine (4-MP) – 4 obtained by the method described in the manufacture WO 97/11939 pamphlet methylene piperidine hydriodic acid salt of (4-MP · HI) – 4 was cooled by stirring in an ice bath under a solution 0.66M 20mL (13.19mmol). Thereafter, the solution was added 57% hydroiodic acid and 2.66g (11.84mmol), and the mixture was stirred for 15 minutes in an ice bath. After concentrated under reduced pressure, to precipitate a white solid by performing twice the operation for azeotropic water and concentrated under reduced pressure and toluene was added to 1.6mL. The washed suspension for 1 hour at room temperature by addition of diisopropyl ether 6mL. Thereafter, The crystals are filtered, washed and crystallized with diisopropyl ether, 4 and dried under reduced pressure at room temperature – was obtained (90% yield) methylene piperidine hydroiodide (4-MP · HI) 2.66g.
1 H-NMR (500MHz, CDCl 3)
δ: 2.66 (4H, t, J = 6.1Hz), 3.31-3.33 (4H, m), 4.91 (2H, s), 8.34 (1H, br s).
Methanol / water mixture methylene piperidine (4-MP) – 4 obtained by the method described in the manufacture WO 97/11939 pamphlet methylene piperidine hydriodic acid salt of (4-MP · HI) – 4 was cooled by stirring in an ice bath under a solution 0.66M 20mL (13.19mmol). Thereafter, the solution was added 57% hydroiodic acid and 2.66g (11.84mmol), and the mixture was stirred for 15 minutes in an ice bath. After concentrated under reduced pressure, to precipitate a white solid by performing twice the operation for azeotropic water and concentrated under reduced pressure and toluene was added to 1.6mL. The washed suspension for 1 hour at room temperature by addition of diisopropyl ether 6mL. Thereafter, The crystals are filtered, washed and crystallized with diisopropyl ether, 4 and dried under reduced pressure at room temperature – was obtained (90% yield) methylene piperidine hydroiodide (4-MP · HI) 2.66g.
1 H-NMR (500MHz, CDCl 3)
δ: 2.66 (4H, t, J = 6.1Hz), 3.31-3.33 (4H, m), 4.91 (2H, s), 8.34 (1H, br s).
Preparation Example 5
The reaction was carried out similarly to the above method by using trifluoroacetic acid (TFA) 1.35g and (11.87mmol) in place of hydriodic acid production 57% methylene piperidine trifluoroacetate salt of (4-MP · TFA), – 4 I got a (92% yield) methylene piperidine trifluoroacetate (4-MP · TFA) 2.55g – 4.
1 H-NMR (500MHz, CDCl 3)
δ: 2.50 (4H, t, J = 6.1Hz), 3.16 (4H, t, J = 6.1Hz), 4.89 (2H, s), 9.52 (1H, br s).
The reaction was carried out similarly to the above method by using trifluoroacetic acid (TFA) 1.35g and (11.87mmol) in place of hydriodic acid production 57% methylene piperidine trifluoroacetate salt of (4-MP · TFA), – 4 I got a (92% yield) methylene piperidine trifluoroacetate (4-MP · TFA) 2.55g – 4.
1 H-NMR (500MHz, CDCl 3)
δ: 2.50 (4H, t, J = 6.1Hz), 3.16 (4H, t, J = 6.1Hz), 4.89 (2H, s), 9.52 (1H, br s).
Preparation Example 6
The reaction was carried out in the same manner as the above-described method using 69% nitric acid 1.08g the (11.87mmol) instead of hydroiodic acid production 57% methylene piperidine nitrate (4-MP · HNO 3), 4 – – 4 methylenepiperidine nitrate I got a (89% yield) (4-MP · HNO 3) 1.87g.
1 H-NMR (500MHz, CDCl 3)
δ: 2.53 (4H, t, J = 6.1Hz), 3.28 (4H, t, J = 6.1Hz), 4.89 (2H, s), 8.85 (1H, br s).
The reaction was carried out in the same manner as the above-described method using 69% nitric acid 1.08g the (11.87mmol) instead of hydroiodic acid production 57% methylene piperidine nitrate (4-MP · HNO 3), 4 – – 4 methylenepiperidine nitrate I got a (89% yield) (4-MP · HNO 3) 1.87g.
1 H-NMR (500MHz, CDCl 3)
δ: 2.53 (4H, t, J = 6.1Hz), 3.28 (4H, t, J = 6.1Hz), 4.89 (2H, s), 8.85 (1H, br s).
Example 1
(2R, 3R) -2 – (2,4 – difluorophenyl) -3 – (4 – methylene-piperidin-1 – yl) -1 – (1H-1, 2,4 – triazol-1 – yl) butan-2 – manufacture ol (KP-103)
(2R, 3R) -2 – (2,4 – difluorophenyl) -3 – (4 – methylene-piperidin-1 – yl) -1 – (1H-1, 2,4 – triazol-1 – yl) butan-2 – manufacture ol (KP-103)
Was stirred while addition of acetonitrile 80mL, lithium hydroxide 2.859g methylene piperidine hydrobromide (4-MP · HBr) 21.26g and (119.4mmol) and (119.4mmol) – 4 obtained in Production Example 1. Then, (2R, 3S) -2 – (2,4 – difluorophenyl) -3 – methyl -2 – [(1H-1, 2,4 - triazol-1 - yl) methyl] oxirane and 20g (79.6mmol) was added, and the mixture was heated under reflux for 14 hours at (external temperature 100 ℃) oil bath. After completion of the reaction, to precipitate the crystals by the addition of ethanol and distilled water to the reaction solution. Thereafter, the crystals were filtered, washed with ethanol / water mixture 40mL, and naturally dried at room temperature for 12 hours and dried under reduced pressure at 40 ℃, KP-103 24.2g light yellow 87.3% (yield, HPLC purity 95.3 % I got).
1 H-NMR (500MHz, CDCl 3)
δ: 0.96 (3H, dd, J = 2.68, 7.08 Hz), 2.13-2.26 (4H, m), 2.35 (2H, br), 2.70 (2H, br) ,2.90-2 .94 (1H, q, J = 7.08 Hz), 4.64 (2H, s), 4.82 (1H, dd, J = 0.73, 14.39 Hz), 4.87 (1H, dd, J = 0.73, 14.39 Hz), 5.45 (1H, s), 6.72-6.81 (2H , m), 7.51 (1H, dt, J = 6.59, 9.03 Hz), 7.78 (1H, s),
8.02 (1H, s).
FAB-MS m / z: 349 [M + H] +
:86-89 ℃ melting point
Optical rotation: [α] D 25 -87 ~ -91 ° (C = 1.0, methanol)
1 H-NMR (500MHz, CDCl 3)
δ: 0.96 (3H, dd, J = 2.68, 7.08 Hz), 2.13-2.26 (4H, m), 2.35 (2H, br), 2.70 (2H, br) ,2.90-2 .94 (1H, q, J = 7.08 Hz), 4.64 (2H, s), 4.82 (1H, dd, J = 0.73, 14.39 Hz), 4.87 (1H, dd, J = 0.73, 14.39 Hz), 5.45 (1H, s), 6.72-6.81 (2H , m), 7.51 (1H, dt, J = 6.59, 9.03 Hz), 7.78 (1H, s),
8.02 (1H, s).
FAB-MS m / z: 349 [M + H] +
:86-89 ℃ melting point
Optical rotation: [α] D 25 -87 ~ -91 ° (C = 1.0, methanol)
Example 2
Epoxy triazole 0.50g (1.99mmol), 4 – in addition to acetonitrile 2mL lithium hydroxide 0.07g methylene piperidine hydrobromide (4-MP · HBr) 0.53g and (2.98mmol) and (2.96mmol), oil bath ( I was heated under reflux for 14 hours at an external temperature of 100 ℃).After the solvent was evaporated under reduced pressure of the reaction solution obtained, the solution was separated by the addition of ethyl acetate and water to the residue. The organic layer was concentrated under reduced pressure and purified by silica gel column chromatography (1:1) hexane / ethyl acetate solvent, to give (86% yield) KP-103 0.59g.
Epoxy triazole 0.50g (1.99mmol), 4 – in addition to acetonitrile 2mL lithium hydroxide 0.07g methylene piperidine hydrobromide (4-MP · HBr) 0.53g and (2.98mmol) and (2.96mmol), oil bath ( I was heated under reflux for 14 hours at an external temperature of 100 ℃).After the solvent was evaporated under reduced pressure of the reaction solution obtained, the solution was separated by the addition of ethyl acetate and water to the residue. The organic layer was concentrated under reduced pressure and purified by silica gel column chromatography (1:1) hexane / ethyl acetate solvent, to give (86% yield) KP-103 0.59g.
Example 3
The reaction was carried out in the same manner as in Example 2 using the calcium hydroxide 0.22g (2.97mmol) instead of lithium hydroxide, to give (82% yield) KP-103 0.57g.
The reaction was carried out in the same manner as in Example 2 using the calcium hydroxide 0.22g (2.97mmol) instead of lithium hydroxide, to give (82% yield) KP-103 0.57g.
Example 4
Was performed for 19 hours and the reaction in the same manner as in Example 2 using strontium hydroxide 0.36g a (2.98mmol) in place of lithium hydroxide, to give (68% yield) KP-103 0.47g.
Was performed for 19 hours and the reaction in the same manner as in Example 2 using strontium hydroxide 0.36g a (2.98mmol) in place of lithium hydroxide, to give (68% yield) KP-103 0.47g.
Example 5
Was added 2mL of acetonitrile lithium hydroxide monohydrate 0.13g methylene piperidine hydrobromide (4-MP · HBr) 0.53g and (2.98mmol) and (2.96mmol) – epoxy-triazole 0.50g (1.99mmol), 4 , I was heated under reflux for 14 hours at (external temperature of 100 ℃) oil bath. Was determined to (relative area percentage of KP-103) reaction rate by HPLC measurements by sampling the reaction mixture, it was confirmed the formation of KP-103 at 81% response rate.
Was added 2mL of acetonitrile lithium hydroxide monohydrate 0.13g methylene piperidine hydrobromide (4-MP · HBr) 0.53g and (2.98mmol) and (2.96mmol) – epoxy-triazole 0.50g (1.99mmol), 4 , I was heated under reflux for 14 hours at (external temperature of 100 ℃) oil bath. Was determined to (relative area percentage of KP-103) reaction rate by HPLC measurements by sampling the reaction mixture, it was confirmed the formation of KP-103 at 81% response rate.
Example 6
The reaction was carried out in the same manner as in Example 2 using cyclopentyl methyl ether (CPME) 2mL instead of acetonitrile, to give (91% yield) KP-103 0.63g.
The reaction was carried out in the same manner as in Example 2 using cyclopentyl methyl ether (CPME) 2mL instead of acetonitrile, to give (91% yield) KP-103 0.63g.
Example 7
1,2 instead of acetonitrile – The reaction was carried out in the same manner as in Example 2 using dimethoxyethane (DME) 2mL, was obtained (79% yield) KP-103 0.55g.
1,2 instead of acetonitrile – The reaction was carried out in the same manner as in Example 2 using dimethoxyethane (DME) 2mL, was obtained (79% yield) KP-103 0.55g.
Example 8
1 in place of acetonitrile – The reaction was carried out in the same manner as in Example 2 using butanol 2mL, was obtained (72% yield) KP-103 0.59g.
1 in place of acetonitrile – The reaction was carried out in the same manner as in Example 2 using butanol 2mL, was obtained (72% yield) KP-103 0.59g.
Example 9
The reaction was carried out in the same manner as in Example 2 using isopropanol 2mL instead of acetonitrile, to give (86% yield) KP-103 0.50g.
The reaction was carried out in the same manner as in Example 2 using isopropanol 2mL instead of acetonitrile, to give (86% yield) KP-103 0.50g.
Example 10
Methyl – 2 – 4 instead of acetonitrile reaction was carried out in the same manner as in Example 2 using the pentanone (MIBK) 2mL, to give (88% yield) KP-103 0.61g.
Methyl – 2 – 4 instead of acetonitrile reaction was carried out in the same manner as in Example 2 using the pentanone (MIBK) 2mL, to give (88% yield) KP-103 0.61g.
Example 11
Example Using methylene piperidine hydrochloride (4-MP · HCl) 0.40g and (2.99mmol) – 4 obtained in Production Example 3 in place of methylene piperidine hydrobromide salt of (4-MP · HBr) – 4 The reaction was carried out in the same manner as 2, was obtained (67% yield) KP-103 0.47g.
Example Using methylene piperidine hydrochloride (4-MP · HCl) 0.40g and (2.99mmol) – 4 obtained in Production Example 3 in place of methylene piperidine hydrobromide salt of (4-MP · HBr) – 4 The reaction was carried out in the same manner as 2, was obtained (67% yield) KP-103 0.47g.
Example 12
Using methylene piperidine hydroiodide (4-MP · HI) 0.67g and (2.99mmol) – 4 obtained in Production Example 4 in place of methylene piperidine hydrobromide salt of (4-MP · HBr) – 4 The reaction was carried out in the same manner as in Example 2 upgrade does not give (90% yield) KP-103 0.62g.
Using methylene piperidine hydroiodide (4-MP · HI) 0.67g and (2.99mmol) – 4 obtained in Production Example 4 in place of methylene piperidine hydrobromide salt of (4-MP · HBr) – 4 The reaction was carried out in the same manner as in Example 2 upgrade does not give (90% yield) KP-103 0.62g.
Example 13
Using methylene piperidine trifluoroacetate (4-MP · TFA) 0.63g and (2.98mmol) – 4 obtained in Production Example 5 in place of methylene piperidine hydrobromide salt of (4-MP · HBr) – 4 The reaction was carried out in the same manner as in Example 2, was obtained (78% yield) KP-103 0.54g.
Using methylene piperidine trifluoroacetate (4-MP · TFA) 0.63g and (2.98mmol) – 4 obtained in Production Example 5 in place of methylene piperidine hydrobromide salt of (4-MP · HBr) – 4 The reaction was carried out in the same manner as in Example 2, was obtained (78% yield) KP-103 0.54g.
Example 14
Example Using methylenepiperidine nitrate (4-MP · HNO 3) 0.48g and (3.00mmol) – 4 obtained in Production Example 6 in place of methylene piperidine hydrobromide salt of (4-MP · HBr) – 4 The reaction was carried out in the same manner as 2, was obtained (71% yield) KP-103 0.49g.
Example Using methylenepiperidine nitrate (4-MP · HNO 3) 0.48g and (3.00mmol) – 4 obtained in Production Example 6 in place of methylene piperidine hydrobromide salt of (4-MP · HBr) – 4 The reaction was carried out in the same manner as 2, was obtained (71% yield) KP-103 0.49g.
Example 15
Methylenepiperidine hydroiodic acid – 4 obtained in Production Example 4 in place of methylene piperidine hydrobromide salt of (4-MP · HBr) – Sodium hydroxide 0.12g (2.98mmol), 4 instead of lithium hydroxide was performed 18 hours and the reaction in the same manner as in Example 2 using salt (4-MP · HI) 0.67g and (2.99mmol), to give (73% yield) KP-103 0.51g.
Methylenepiperidine hydroiodic acid – 4 obtained in Production Example 4 in place of methylene piperidine hydrobromide salt of (4-MP · HBr) – Sodium hydroxide 0.12g (2.98mmol), 4 instead of lithium hydroxide was performed 18 hours and the reaction in the same manner as in Example 2 using salt (4-MP · HI) 0.67g and (2.99mmol), to give (73% yield) KP-103 0.51g.
Conventional methods for producing formula 1 compound of the starting material 4 – impurity contamination at the acquisition stage of methylene piperidine, and by-products to be produced during the manufacture of the formula 1 compound was a problem. In the method of the present invention, as a starting material of the production method of the formula 1 compound, 4 – by making the acid addition salt of methylene-piperidine, 4 – impurities incorporated in the acquisition phase of the methylene piperidine has been removed, high purity it is possible to use a solid. In the method of the present invention, since the ring-opening addition of the amine to epoxy triazole is promoted, 4 – there is no need to use excess methylene piperidine, high yield, and by-products of the compound of Formula 1 under mild conditions It can be produced by reducing compound. Therefore, by the method of the present invention to produce an industrial scale formula 1 compound became possible.
Patent Citations
| CITED PATENT | FILING DATE | PUBLICATION DATE | APPLICANT | TITLE |
|---|---|---|---|---|
| WO1994026734A1 | May 2, 1994 | Nov 24, 1994 | Tadashi Arika | Azolylamine derivative |
| WO1997011939A1 | Sep 26, 1996 | Apr 3, 1997 | Kaken Pharma Co Ltd | Process for the preparation of 4-methylenepiperidines |
| JPH02191262A | Title not available |
Non-Patent Citations
| REFERENCE | ||
|---|---|---|
| 1 | EUR. J. ORG. CHEM. 2004, pages 3597 – 3600 | |
| 2 | J. ORG. CHEM. vol. 72, 2007, pages 3713 – 3722 | |
| 3 | * | MIMURA, MITSUO ET AL.: ‘Synthesis and evaluation of (piperidinomethylene)bis (phosphonic acid) derivatives as anti- osteoporosis agents‘ CHEMICAL & PHARMACEUTICAL BULLETIN vol. 41, no. 11, 1993, pages 1971 – 1986 |
| 4 | * | OGURA, HIRONOBU ET AL.: ‘Synthesis and antifungal activities of (2R,3R)-2-aryl-1-azolyl-3-(substituted amino)-2-butanol derivatives as topical antifungal agents‘ CHEMICAL & PHARMACEUTICAL BULLETIN vol. 47, no. 10, 1999, pages 1417 – 1425, XP002296880 |
| 5 | SYNTHESIS 2004, pages 1563 – 1565 | |
PATENTS
10-6-1999
|
Azolylamine derivative
| |
2-11-1998
|
Azolylamine derivative
| |
12-18-1997
|
AZOLYLAMINE DERIVATIVE
| |
4-16-1997
|
Azolylamine derivative
|
References
- Patel T, Dhillon S (Nov 2013). “Efinaconazole: first global approval”. Drugs 73 (17): 1977–1983. doi:10.1007/s40265-013-0152-x.PMID 24249649.
- Tschen EH, Bucko AD, Oizumi N, Kawabata H, Olin JT, Pillai R (Feb 2013). “Efinaconazole solution in the treatment of toenail onychomycosis: a phase 2, multicenter, randomized, double-blind study”. J Drugs Dermatol 12 (2): 186–192. PMID 23377392.
- Tatsumi Y, Nagashima M, Shibanushi T, et al. (May 2013). “Mechanism of action of efinaconazole, a novel triazole antifungal agent”. Antimicrob Agents Chemother 57 (5): 2405–2509.
8 ALBACONAZOLE
Albaconazole
Also known as: UNII-YDW24Y8IAB; UR-9825; 187949-02-6; UR 9825, W-0027
Molecular Formula: C20H16ClF2N5O2 Molecular Weight: 431.823146
(1R,2R)-7-chloro-3-[2-(2,4-difluorophenyl)-2-hydroxy-1-methyl-3-(1H-1,2,4-triazol-1-yl)propyl]quinazolin-4(3H)-one
7-chloro-3-[(2R,3R)-3-(2,4-difluorophenyl)-3-hydroxy-4-(1,2,4-triazol-1-yl)butan-2-yl]quinazolin-4-one
Albaconazole is a broad-spectrum antifungal agent being evaluated in phase II clinical trials by Stiefel for the oral treatment of fungal infections, including toenail fungus, distal onychomycosis and subungual onychomycosis. Early clinical trials for the treatment of tinea pedis have been completed. In September 2005, Uriach, originator of albaconazole, granted Stiefel exclusive rights to develop and market albaconazole on a worldwide basis. In November 2006, Uriach’s R&D pipeline was transferred to Palau Pharma, a newly-created spin-out company. Under the terms of the agreement with Stiefel, Palau retains rights as comarketing partner in some European countries. In August 2013, Palau Pharma granted worldwide rights to Actavis. A triazole, albaconazole, has shown potent activity against a broad range of organisms, including pathogens resistant to other antifungals, such as fluconazole or itraconazole. It will be developed as an oral and topical formulation, and is expected to be available to the medical community for a variety of dermatological indications and fungal infections, including vulvovaginal candidiasis.
 | |
| SYSTEMATIC (IUPAC) NAME | |
|---|---|
| 7-Chloro-3-[(2R,3R)-3-(2,4-difluorophenyl)-3-hydroxy-4-(1,2,4-triazol-1-yl)butan-2-yl]quinazolin-4-one | |
| CLINICAL DATA | |
| IDENTIFIERS | |
| CAS NUMBER | 187949-02-6 |
| ATC CODE | None |
| PUBCHEM | CID 208952 |
| CHEMSPIDER | 181045 |
| UNII | YDW24Y8IAB |
| KEGG | D09702 |
| CHEMBL | CHEMBL298817 |
| CHEMICAL DATA | |
| FORMULA | C20H16ClF2N5O2 |
| MOL. MASS | 431.823146 g/mol |
11-26-2003
|
Method for preparing pyrimidone derivatives with antifungal activity
|
The condensation of the chiral oxazolidinone (I) with 2,4-difluorophenacyl bromide (II) by means of NaHMDS in THF/Et2 O gives the chiral oxirane (III), which is treated with LiOH and H2O2 to eliminate the chiral auxiliary, yielding the carboxylic acid (IV). The cleavage of the oxirane ring of (IV) with 1,2,4-triazole (V) and NaH in hot DMF affords the chiral hydroxyacid (VI), which is submitted to Curtius rearrangement by means of DPPA in hot pyridine to provide the chiral oxazolidinone (VII). The cleavage of the oxazolidinone ring of (VII) by means of refluxing aq. HCl gives the chiral aminoalcohol (VIII), which is condensed with 2-amino-4-chlorobenzoic acid (IX) by means of DCC and HOBt to yield the corresponding amide (X). Finally, this compound is cyclized to the target quinazolinone by reaction with triethyl orthoformate in hot dioxane/NMP.
The condensation of the chiral oxazolidinone (I) with 2,4-difluorophenacyl bromide (II) by means of NaHMDS in THF/Et2 O gives the chiral oxirane (III), which is treated with LiOH and H2O2 to eliminate the chiral auxiliary, yielding the carboxylic acid (IV). The cleavage of the oxirane ring of (IV) with 1,2,4-triazole (V) and NaH in hot DMF affords the chiral hydroxyacid (VI), which is submitted to Curtius rearrangement by means of DPPA in hot pyridine to provide the chiral oxazolidinone (VII). The cleavage of the oxazolidinone ring of (VII) by means of refluxing aq. HCl gives the chiral aminoalcohol (VIII), which is condensed with 2-amino-4-chlorobenzoic acid (IX) by means of DCC and HOBt to yield the corresponding amide (X). Finally, this compound is cyclized to the target quinazolinone by reaction with triethyl orthoformate in hot dioxane/NMP.
EP 0783501; ES 2107376; ES 2120885; JP 1998508317; US 5807854; WO 9705130
…………………………………………..
The condensation of (R)-lactic acid (I) with morpholine (II) gives the corresponding morpholide (III), which is protected at the hydroxyl position with dihydropyran (IV) to yield the tetrahydropyranyl ether (V). The Grignard reaction of (V) with 2,4-difluorophenylmagnesium bromide (VI) affords the chiral 1-propanone (VII), which by a Corey’s diastereoselective epoxidation with trimethylsulfoxonium iodide is converted into the oxirane (VIII). The opening of the oxirane ring of (VIII) by means of 1,2,4-triazole (IX) and NaH provides the tertiary alcohol (X), which is treated with pyridine p-toluenesulfonate to give the deprotected diol (XI) as a (2R,3R) and (2R,3S) 4:1 diastereomeric mixture, from which the desired (2R,3R)-isomer (XII) was isolated by crystallization. The reaction of (XII) with Ms-Cl and TEA, followed by cyclization with NaOMe, yields the oxirane (XIII), which is finally condensed with 7-chloroquinazolin-4(3H)-one (XIV) by means of K2CO3 in hot NMP.
ES 2159488; WO 0166519
…………………………………………….
Alternatively, intermediate (XIII) can be obtained as follows: Heating of ethyl (S)-lactate (XIV) with morpholine affords amide (XVI), which then reacts with 3,4-dihydro-2H-pyran (A) in the presence of p-TsOH to give protected derivative (XVII). Grignard reaction between (XVII), bromo derivative (XVIII) and Mg turnings in THF yields protected ketone (XIX), which is treated with pyridinium p-toluenesulfonate (PPTS) (THP group removal) and reprotected by means of Tf2O and DIEA to give triflate derivative (XX). Conversion of (XX) into intermediate (XIII) is achieved by reaction with triazolone (VII) and NaH in THF.
Chem Pharm Bull 1993,41(6),1035-42
……………………………………
Alternatively, derivative (XXIX) can be obtained in an analogous way as its enantiomer (XIX). Diastereoselective epoxidation of (XXIX) with trimethylsulfoxonium iodide and NaH in DMSO provides oxirane (XXX) (3). THP group removal by means of PPTS in EtOH, followed by reaction with 3,5-dinitrobenzoyl chloride (XXXI) and NaHCO3 in CH2Cl2, yields a diastereomeric mixture from which dinitrobenzoate derivative (2R,3R)-(XXXII) is obtained by recrystallization (1). Hydrolysis of (2R,3R)-(XXXII) in MeOH by treatment with aqueous NaOH gives compound (2R,3R)-(XXXIII), which is converted into ester (2R,3S)-(XXXIV) by Mitsunobu reaction with benzoic acid, Ph3P and DEAD in THF. Subsequent debenzoylation of (2R,3S)-(XXXIV) with NaOMe in MeOH affords oxiranyl ethanol derivative (2R,3R)-(XXXV), which is first converted into its triflate derivative by means of Tf2O and DIEA in CH2Cl2, and then into triazolone derivative (2S,3R)-(XXXVI) by reaction with intermediate (VII) and NaH in CH2Cl2/DMF. Finally, oxirane derivative (2S,3R)-(XXXVI) reacts with triazole (XXVI) and NaH in DMF to furnish the desired product.
…………………………………………………..
ER-30346 is synthesized by thiazole ring formation of (2R,3R)-3-(2,4-difluorophenyl)-3-hydroxy-2-methyl-4-(1H-1,2,4-triazol-1-yl)thiobutanamide (I) and 4-bromoacetylbenzonitrile (II) by means of reflux in methanol. The thioamide (I) is obtained with excellent yield from a chiral nitrile (III) by heating with diethyl dithiophosphate in aqueous medium.
…………………………………………….
The nitrile (III), a chiral key intermediate of this synthesis, can be obtained by two different synthetic routes as follows: Route-a: The key step of this route is ring opening reaction of the trisubstituted oxirane (VII) by cyanide anion leading to the nitrile (III). The chiral oxirane (VII) is synthesized from (R)-lactic acid derivatives as already reported. The reaction of (VII) with diethylaluminum cyanide in toluene or lithium cyanide in tetrahydrofuran gives the nitrile (III) with high yield without any epimerization reaction.
…………………………………………..
The nitrile (III), a chiral key intermediate of this synthesis, can be obtained by two different synthetic routes as follows: Route-b: The starting material of this route is methyl (S)-3-hydroxy-2-methylpropionate (VIII), which contains one additional carbon between the hydroxyl group and the 2-position carbon of (R)-lactate, the starting material of route-a. The hydroxyl group of (VIII) is protected by triphenylmethyl group. Then, 2,4-difluorophenyl moiety is introduced to give the ketone (X). Direct conversion of the ketone (X) to the oxirane (XIV) by dimethylsulfoxonium methylide, the same condition for compound (IV) in route-a, does not proceed. The oxirane (XIV) having desired stereochemistry is obtained via oxidation reaction. The ketone (X) is converted to the exomethylene (XI) by Wittig reaction. The stereoselective oxidation of (XI) is achieved by means of osmium tetroxide in the presence of 4-methylmorpholine N-oxide to give the diol (XII) in 58% yield after separation of its epimer by column chromatography. After methanesulfonylation of the primary alcohol of (XII), a triazole moiety is introduced and the triphenylmethyl group is deprotected. Then, the primary hydroxyl group of (XVI) is oxidized under Swern oxidation condition to give the aldehyde (XVII), which is converted to the chiral nitrile intermediate (III) by means of heating with hydroxylamine-O-sulfonic acid.
………………………………..
J. Med. Chem., 1998, 41 (11), pp 1869–1882
DOI: 10.1021/jm9707277
A series of azole antifungal agents featuring a quinazolinone nucleus have been subjected to studies of structure−activity relationships. In general, these compounds displayed higher in vitro activities against filamentous fungi and shorter half-lives than the structures described in our preceding paper. The most potent products in vitro carried a halogen (or an isostere) at the 7-position of the quinazolinone ring. Using a murine model of systemic candidosis, oral activity was found to be dependent on hydrophobicity, which, in turn, modulated the compound’s half-life. The 7-Cl derivative, (1R,2R)-7-chloro-3-[2-(2,4-difluorophenyl)-2-hydroxy-1-methyl-3-(1H-1,2,4-triazol-1-yl)propyl]quinazolin-4(3H)-one (20, UR-9825), was selected for further testing due to its high in vitro activity, low toxicity, good pharmacokinetic profile, and ease of obtention. Compound 20 is the (1R,2R) isomer of four possible stereoisomers. The other three isomers were also prepared and tested. The enantiomer (1S,2S) and the (1R,2S) epimer were inactive, whereas the (1S,2R) epimer retained some activity. In vitro 20 was superior to fluconazole, itraconazole, SCH-42427, and TAK-187 and roughly similar to voriconazole and ER-30346. In vivo, 20 was only moderately active in a mouse model of systemic candidosis when administration was limited to the first day. This was attributed to its short half-life in that species (t1/2 = 1 h po). Protection levels comparable to or higher than those of fluconazole, however, were observed in systemic candidosis models in rat and rabbit, where the half-life of the compound was found to be 6 and 9 h, respectively. Finally, 20 showed excellent protection levels in an immunocompromised rat model of disseminated aspergillosis. The compound showed low toxicity signs when administered to rats at 250 mg/kg qd or at 100 mg/kg bid during 28 days.
The condensation of the chiral oxazolidinone (I) with 2,4-difluorophenacyl bromide (II) by means of NaHMDS in THF/Et2 O gives the chiral oxirane (III), which is treated with LiOH and H2O2 to eliminate the chiral auxiliary, yielding the carboxylic acid (IV). The cleavage of the oxirane ring of (IV) with 1,2,4-triazole (V) and NaH in hot DMF affords the chiral hydroxyacid (VI), which is submitted to Curtius rearrangement by means of DPPA in hot pyridine to provide the chiral oxazolidinone (VII). The cleavage of the oxazolidinone ring of (VII) by means of refluxing aq. HCl gives the chiral aminoalcohol (VIII), which is condensed with 2-amino-4-chlorobenzoic acid (IX) by means of DCC and HOBt to yield the corresponding amide (X). Finally, this compound is cyclized to the target quinazolinone by reaction with triethyl orthoformate in hot dioxane/NMP.
J Med Chem 1998,41(11),1869
(1R,2R)-7-Chloro-3-[2-(2,4-difluorophenyl)-2-hydroxy-1-methyl-3-(1H-1,2,4-triazol-1-yl)propyl]quinazolin-4(3H)-one (20, UR-9825). Precipitated from EtOH/H2O (66% yield from amine 11): white amorphous solid;
mp 93−110 °C (wide range);
IR (KBr) ν 1675, 1601, 1554, 1498 cm-1;
1H NMR (300 MHz, CDCl3) 8.58 (s, 1H, N CH-N), 8.26 (d, J = 8.6, 1H, arom), 8.11 (d, J = 5.7, trace rotamer), 7.76 (s, 2H, triazol),
CH-N), 8.26 (d, J = 8.6, 1H, arom), 8.11 (d, J = 5.7, trace rotamer), 7.76 (s, 2H, triazol),
CH-N), 8.26 (d, J = 8.6, 1H, arom), 8.11 (d, J = 5.7, trace rotamer), 7.76 (s, 2H, triazol),
7.74 (d, J = 5.3, 1H, arom), 7.5 (m, 2H, arom), 7.10 (s, trace rotamer), 6.9−6.7 (m, 2H, arom),
5.91 (dq, Jd = 2, Jq = 7, 1H, MeCH), 5.54 (d, J = 2, 1H, OH),
5.15 (d, J = 14.2 1H, CH(H)), 4.9−4.7 (m, trace rotamer), 4.30 (d, trace rotamer), 3.99 (d, J = 14.2, 1H, CH(H)),
1.46 (d, J = 6.9, trace rotamer), 1.29 (d, J = 7, 3H, CHMe);
GC−MS 224 (Tr-CH2COHAr, C10H8F2N3O), 208 (group N-ethylheterocycle, C10H9ClN2O);
[α]D −8.0° (c 1, CHCl3).
Chiral HPLC (column, CicloBond SN 1; eluent, MeOH: Et3NHOAc in H2O at pH7 1:1; retention times: (S,S) (74) tR 12.6 min; (R,R) (20) tR 13.7 min). Area ratio: 0.01:99.99.
Anal. (C20H16ClF2N5O2) C, H, N.
9 BUTOCONAZOLE












SEE PART 2 AT http://apisynthesisint.blogspot.in/p/blog-page.html

1-(4-(4-chlorophenyl)-2-(2,6-dichlorophenylthio)-n-butyl)-1H-imidazole
64872-77-1 NITRATE ,
64872-76-0 (free base)
Butoconazole nitrate, RS-35887-00-10-3, RS-35887, Gynomyk, Gynazole-1, Femstat
1-[4-(4-Chlorophenyl)-2-[(2,6-dichlorophenyl)thio]butyl]-1H-imidazole
Molecular Formula: C19H17Cl3N2S
Molecular Weight: 411.78
Percent Composition: C 55.42%, H 4.16%, Cl 25.83%, N 6.80%, S 7.79%
Properties: Crystals from cyclohexane, mp 68-70.5°.
Melting point: mp 68-70.5°
Derivative Type: Nitrate
CAS Registry Number: 64872-77-1
Manufacturers’ Codes: RS-35887
Trademarks: Femstat (Syntex); Gynomyk (Cassenne)
Molecular Formula: C19H17Cl3N2S.HNO3
Molecular Weight: 474.79
Percent Composition: C 48.06%, H 3.82%, Cl 22.40%, N 8.85%, S 6.75%, O 10.11%
Properties: Colorless blades from acetone/ethyl acetate, mp 162-163°. LD50 in mice, male, female rats (mg/kg): >3200, >3200, 1720 orally; >1600, 940, 940 i.p. (Walker).
Melting point: mp 162-163°
Toxicity data: LD50 in mice, male, female rats (mg/kg): >3200, >3200, 1720 orally; >1600, 940, 940 i.p. (Walker)
Therap-Cat: Antifungal (topical).
Butoconazole (trade names Gynazole-1, Mycelex-3) is an imidazoleantifungal used in gynecology. It is administered as a vaginal cream.[1][2]
For the local treatment of vulvovaginal candidiasis (infections caused by Candida)
Brief background information
| SALT | ATC | FORMULA | MM | CAS |
|---|---|---|---|---|
| - | G01AF15 | C 19 H 17 Cl 3 N 2 S | 411.78 g / mol | 64872-76-0 |
| mononitrate | G01AF15 | C 19 H 17 Cl 3 N 2 S ⋅ HNO 3 | 474.80 g / mol | 64872-77-1 |
No Exclusivity found
| DRUG NAME | Femstat 3 (from Drugs@FDA) |
|---|---|
| ACTIVE INGREDIENT | Butoconazole nitrate |
| DOSAGE FORM | Cream |
| ROUTE | Vaginal |
| STRENGTH | 2% |
| MARKET STATUS | Over the Counter |
| COMPANY | Bayer |
- 64872-77-1 , butoconazole nitrate
- CAS Number: 64872-77-1
- Molecular Weight: 474.79
- Molecular Formula: C19H17Cl3N2S ·HNO3
| PATENT NO | PATENT EXPIRY | |
|---|---|---|
| 5993856 | Nov 17, 2017 |
Laszlo Czibula, Laszlo Dobay, Eva Werkne Papp, Judit Nagyne Bagdy, Ferenc Sebok, “High Purity Butoconazole Nitrate with Specified Particle Size and a Process for the Preparation Thereof.” U.S. Patent US20080221190, issued September 11, 2008.
 | |
| SYSTEMATIC (IUPAC) NAME | |
|---|---|
| 1-[4-(4-Chlorophenyl)-2-(2,6-dichlorophenyl)sulfanylbutyl]imidazole | |
| CLINICAL DATA | |
| TRADE NAMES | Gynazole-1, Mycelex-3 |
| AHFS/DRUGS.COM | monograph |
| MEDLINEPLUS | a682012 |
| PREGNANCY CAT. |
|
| LEGAL STATUS | |
| ROUTES | Vaginal cream |
| IDENTIFIERS | |
| CAS NUMBER | 67085-13-6 |
| ATC CODE | G01AF15 |
| PUBCHEM | CID 47472 |
| DRUGBANK | DB00639 |
| CHEMSPIDER | 43192 |
| UNII | 0Q771797PH |
| KEGG | D00880 |
| CHEBI | CHEBI:3240 |
| CHEMBL | CHEMBL1295 |
| CHEMICAL DATA | |
| FORMULA | C19H17Cl3N2S |
| MOL. MASS | 411.776 g/mol |
Use
- an antifungal agent for topical use
Classes substance
- Eter chlorothiophenol
- Imidazoles
Synthesis pathway
| SYNTHESIS OF A) |
|---|
  |
Trade names
| COUNTRY | TRADE NAME | MANUFACTURER |
|---|---|---|
| France | Ginomik | Cassenne |
| USA | Femstat | Syntex |
| Ukraine | Gіnofort | BAT “Gideon Rіhter” Ugorschina |
Formulations
- 2% vaginal cream
Reference for syn
- Synthesis of a)
- Walker, KAM et al .: J. Med. Chem. (JMCMAR) 21, 840 (1978).
- US 4,078,071 (Syntex; USA-prior. 28.7.1975).
- DOS 2,800,755
………………………
Patent
Butoconazole nitrate (chemical name: l-[4-(4-chlorophenyl)-2-(2,6-dichloro- -phenylthio)-n-butyl]-imidazol nitrate) is a compound of the formula (I),

(I)
it belongs among the aryl-ethylimidazole compounds, has fungicidal activity and may be used for the treatment of vaginal infections caused primarily by Candida albicans. Azoles exert their antifungal effect via modifying the ergosterol synthesis of fungus cells; more particularly, imidazoles inhibit the 14α-demethylase enzyme, thereby bringing about an increased level of 14α-methyl sterols which, in turn, causes an alteration of cell membrane permeability leading to the destruction of the fungus cells (Tetrahedron: Asymmetry Vol 4, No. 7, pp. 1521-1526, 1993). The first process for the preparation of the butoconazole nitrate is a multistep synthesis disclosed in the US 4,078,071 patent specification. Here two reaction routes are given for the preparation of the key intermediate of the formula (TV) (l-[4-(4-chlorophenyl)-2-hydroxy-n- -butyl] -imidazole) .

(IN)
According to one of them first an epoxy compound is prepared from an aromatic aldehyde or from an olefinic compound having a terminal double bond; then the epoxy compound is reacted with imidazole to yield the key intermediate. The aromatic aldehyde (VIII)

(VIII)
is treated with expensive and hazardous reagents (trimethylsulfoxonium iodide and sodium hydride) in dry dimethyl sulfoxide and the epoxide formed in the reaction is isolated after a complicated work-up. The epoxide so obtained is converted to the imidazole derivate in a time consuming reaction in the presence of dimethylformamide, then the key intermediate of the formula (IN) (l-[4-(4-chlorophenyl)-2-hydroxy-n-butyl]-imidazole) is isolated and purified in an additional step. From the compounds having terminal double bond (Nil)

(Nil) the epoxide is obtained via a highly explosive peracidic oxidation step and the epoxide is then converted into (l-[4-(4-chlorophenyl)-2-hydroxy-n-butyl]-imidazole) (IV) in a manner described above. In the other reaction route a poisoning aromatic α-halo-keto compound is used as starting material which is reacted with imidazole to give the corresponding keto-imidazole which, in turn, is reduced with a complex metal hydride – a reagent with potential hazards – to yield the key intermediate (IN). The reaction mixture is worked up in an involved manner. The synthesis way described in J. Med. Chem., 1978, Vol. 21, No. 8, pp 840-843 is as follows: l-chloro-4-chlorophenyl-2-butanol (II)

(II) is treated with the imidazole (III)

(HI)
in the presence of sodium hydride reagent in dimethylformamide solvent. This substitution reaction takes a long time and gives the (l-[4-(4-chlorophenyl)-2-hydroxy-n-butyl]- imidazole) (IN) with a poor yield (51.7 %). In the next step of the butoconazole nitrate synthesis
(l-[4-(4-chlorophenyl)-2-hydroxy-n-butyl]-imidazole) (IN) is treated with thionyl chloride (which is at once a reagent and a solvent) at 65-70 °C to yield l-[4-(4-chlorophenyl)-2-chloro- -n-butyl] -imidazole of the formula (N).

(V)
The reaction mixture is then evaporated to dryness. The removal of the excess thionyl chloride, a highly corrosive substance, requires special equipment; the same applies to waste treatment, an operation which also involves an environmental risk. The residue is dissolved in dichloromethane, the solution is made alkaline by adding aqueous potassium carbonate solution. Phases are separated, the organic layer is washed with water, dried on magnesium sulphate and evaporated to give l-[4-(4-chlorophenyl)-2-chloro-n-butyl]-imidazole (N), as a gum. Said gum is dissolved in acetone and reacted with 2,6-dichlorothiophenol in the presence of potassium carbonate with a long reaction time. After the reaction has been finished, the inorganic salts are removed by filtration, the solvent is evaporated, and the residue is partitioned between water and ether. Butoconazole nitrate is precipitated with nitric acid from the ethereal layer. The end-product crystals in white plates from a mixture of acetone and ethyl acetate (yield: 84 %). Our aim was to provide a process by which the active agent can be prepared in high purity via reaction steps producing good yields and besides that said steps require neither solvents that are highly flammable and explosive (ether), carcinogenic (dimethylformamide) or corrosive (thionylchloride), nor reagents (e. g. sodium hydride) that are highly flammable or explosive. We have surprisingly found that when the starting material l-chloro-4-chlorophenyl-2-
-butanol (II) is reacted with the imidazole (III) in a mixture of toluene and aqueous sodium hydroxide solution in the presence of a phase transfer catalyst, the
(l-[4-(4-chlorophenyl)-2-hydroxy-n-butyl]-imidazole) (IN) key intermediate is obtained with short reaction time and excellent yield (95 %). Next we studied alternative solvents to replace the thionyl chloride in solvent function in the reaction step converting (l-[4-(4-chlorophenyl)-2-hydroxy-n-butyl]-imidazole) (IN) into (l-[4-(4-chlorophenyl)-2-chloro-n-butyl]-imidazole) (N). In the inert solvents which could be taken into account such as dichloromethane, toluene, chlorobenzene and dimethylformamide, the chlorinating reaction yielded a sticky reaction mixture which couldn’t be processed. We have surprisingly found, however that when (l-[4-(4-chlorophenyl)-2-hydroxy-n-butyl]-imidazole) (IN) is dissolved in 1 ,2-dichloroethane and reacted with approximately equimolar amount of thionyl chloride reagent in the presence of catalytic amount of dimethylformamide at 30-35 °C temperature, a crystal suspension is obtained which is easy-to-stir during the whole reaction time, resulting in that chlorination proceeds completely giving l-[4-(4-chlorophenyl)-2-chloro-n-butyl]-imidazole (N) in quantitative yield. Being the compound sufficiently pure, it is not isolated, but separated by extraction and reacted directly with 2,6-dichlorothiophenol in methyl isobutyl ketone to give 1 -[4-(4-chlorophenyl)-2-(2,6-dichlorophenylthio)-n-butyl]-imidazole (VI) (butoconazole).

(NI)
Example 1. Preparation of (1 4-(4-chlorophenyl)-2-hvdroxy-n-butyll-imidazole) (IV) To a solution of 56.7 g (0.26 mol) of l-chloro-4-chloroρhenyl-2-butanol (J. of Medicinal Chemistry, 1978. Nol. 21. No. 8. p. 842) in 200 ml of toluene 36.2 g (0.9 mol) of sodium hydroxide dissolved in 100 ml of water, 6.4 g (0.028 mol) of benzyltriethyammom‘um chloride and 35.2 g (0.51 mol) of imidazole (III) are added. The reaction mixture is heated at 93-95 °C for one hour then the temperature is returned to about 60 °C, the phases are separated and to the organic layer water (100 ml) is added. The mixture is first stirred at 22-25 °C for 1 hour then at 0-5 °C for two hours. The crystals are separated by filtration, washed with water (2 x 35 ml) of 0-5 °C to yield 74 g of wet (l-[4-(4-chloroρhenyl)-2-hydroxy-n-butyl]-imidazole) which is dried at maximum 50 °C in vacuo to give 61.6 g (95 %) of the product. Recrystallization from ethyl acetate gives 52.4 g (85 %) of dry product melting at 104-106 °C.
Example 2. Preparation of l-[4-(4-chlorophenvπ-2-(2,6-(McMorophenyl o)-n-butyl1-ϊmidazole nitrate (I) 25 g (0.1 mol) of l-[4-(4-chlorophenyl)-2-hydroxy-n-butyl]-imidazole (IN) is suspended in 1,2-dichloroethane (125 ml), to this suspension dimethylformamide (1 ml) and thionyl chloride (13.6 g; 0.11 mol) are added at 30-32 °C and the reaction mixture is kept at 35-38 °C for 1.5 hour under stirring. After the chlorination has been finished the homogenous solution is cooled to 15-18 °C, the excess of thionyl choride is decomposed with water (10 ml) then again water (80 ml) is added to the solution. After stirring at 20-22 °C for 0.5 hour the phases are separated and the organic layer is extracted with water (30 ml). To the aqueous solution methyl isobutyl ketone (250 ml) is added and the pH of the mixture is adjusted to 8.5 – 9 with 15 g (0.14 mol) of sodium carbonate dissolved in water (70 ml). The mixture is stirred at 22-25 °C for 0.5 hour, phases are separated, from the organic layer an 50 ml portion is distilled off to remove water and to the remaining solution 26.8 g (0.15 mol) of 2,6-dichloro-thiophenol and 40 g (0.29 mol) of dry potassium carbonate are added. The suspension is stirred at 105 – 108 °C under nitrogen for 3-4 hours. After the reaction has been finished the inorganic salts are removed by filtration at 22-25 °C, the filtrate is washed and clarified with activated carbon and the pH of the clear solution is adjusted to 3 – 3.5 by adding about 8 – 9 ml of 65 % nitric acid. The solution is stirred at the same temperature for 1 hour then the temperature is lowered to 8 – 12 °C. The crystals obtained are filtered and washed to give 48 g of wet l-[4-(4-chlorophenyl)-2-(2,6-dichlorophenylthio)-n-butyl]- -imidazole nitrate corresponding to 42.6 g (90 %) of dry product.
HPLC
Details of the HPLC method: Type of the apparatus: Spectra System/TSP (manufacturer: Thermo Separation Products, USA) Column: LiChrospher RP-18, 250×4.0 mm ID., 5 μm (Merck, Germany, Cat. No. : 1.50983) Mobile phase: methanol : buffer = 8:2 Bujfer: 2.18 g KH2PO4 + 4.18 g K2HPO4-3H2O dissolved in 1000 ml of distilled water; MeOH (HPLC Gradient grade, Merck, Germany, Cat. No.: 1.06007.2500) KH2PO4 (p.a., Merck, Germany, Cat. No.: 1.04877.1000) K2HPO4-3H2O (p.a., Merck, Germany, Cat. No.: 1.05099.1000) Flow rate: 1.0 ml/min Temperature: 40 °C Detection: UN 229 nm Solvent for sampling: eluent Sample concentration: 1.0 mg/ml Injected volume: 10 μl Duration of analysis: 40 min Evaluation: area normalization method. Approximative retention time: 11.9 min B. Particle size: Particle size was determined by sieve analysis using an Alpine sieve operated by a jet of air.
……………………..
WALKER K A M ET AL: “1-[4-(4-Chlorophenyl)-2-(2,6-dichloro phenylthio)-n-butyl]-1H-imidazole nitrate, a new potent antifungal agent” JOURNAL OF MEDICINAL CHEMISTRY, vol. 21, no. 8, August 1978 (1978-08), pages 840-843,
1- [4-(4-chlorophenyl)-2-(2,6-dichlorophenylthio)-n-b~-
tyll-lH-imidazole nitrate (I).
tyll-lH-imidazole nitrate (I).
I as colorless blades
(9.6 g, 84%): mp 162-163 “C (foaming). Anal. (C19H18C13N303S)
C, H, N. The free base prepared by neutralization of a suspension
of 1 in ether with aqueous potassium carbonate and recrystallization
from cyclohexane had mp 68-70.5 “C (foaming).
(9.6 g, 84%): mp 162-163 “C (foaming). Anal. (C19H18C13N303S)
C, H, N. The free base prepared by neutralization of a suspension
of 1 in ether with aqueous potassium carbonate and recrystallization
from cyclohexane had mp 68-70.5 “C (foaming).
……………….
FULL SYNTHESIS
SEE
The chlorohydrin (II) is obtained by the reaction of p-chlorobenzylmagnesium chloride (I) with epichlorohydrin (A) in ether. This is then converted to the crystalline alcohol (III) by reaction with sodium imidazole (B) in DMF. On treatment with thionyl chloride is converted to the corresponding chloro compound (IV). When (IV) is reacted with 2,6-dichloro thiophenol (C) in the presence of anhydrous potassium carbonate in acetone, the free base of butoconazole is formed. Neutralization of the free base (V) with nitric acid gives butoconazole.
References
- Seidman, L. S.; Skokos, C. K. (2005). “An evaluation of butoconazole nitrate 2% site release vaginal cream (Gynazole-1) compared to fluconazole 150 mg tablets (Diflucan) in the time to relief of symptoms in patients with vulvovaginal candidiasis”. Infectious diseases in obstetrics and gynecology 13 (4): 197–206. doi:10.1080/10647440500240615.PMC 1784583. PMID 16338779.
- Butoconazole monograph
Literature References:
Imidazole derivative with antifungal properties. Prepn: K. A. M. Walker, US4078071 (1978 to Syntex).
Prepn, toxicity, activity vs Candida albicans in mice: K. A. M. Walker et al., J. Med. Chem. 21, 840 (1978).
In vitro comparison with other antifungal agents: F. C. Odds et al., J. Antimicrob. Chemother. 14, 105 (1984).
Clinical trials in treatment of vulvovaginal candidiasis: W. Droegemueller et al.,Obstet. Gynecol. 64, 530 (1984); J. B. Jacobson et al., Acta Obstet. Gynecol. Scand. 64, 241 (1985).
Comparison with miconazole, q.v.: C. S. Bradbeer et al., Genitourin. Med. 61,270 (1985).
SEE PART 2 AT http://apisynthesisint.blogspot.in/p/blog-page.html
//////////////////
No comments:
Post a Comment