ZOLID SERIES
1 TEDIZOLID/TOREZOLID
2 RADEZOLID
3 CADAZOLID
4 SUTEZOLID
5 EPEREZOLID
6 MRX1
7 LCB01-0371
8 Ranbezolid
9 FYL 67
10 WCK ?
11
12
under construction

1 TEDIZOLID








 38 nos
38 nos



WILL BE UPDATED

 Eperezolid
Eperezolid
 radezolid
radezolid

 Sutezolid
Sutezolid
 linezolid
linezolid

1 TEDIZOLID/TOREZOLID
2 RADEZOLID
3 CADAZOLID
4 SUTEZOLID
5 EPEREZOLID
6 MRX1
7 LCB01-0371
8 Ranbezolid
9 FYL 67
10 WCK ?
11
12
under construction
1 TEDIZOLID
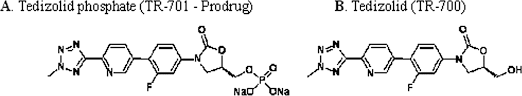
TEDIZOLID PHOSPHATE
[(5R)-3-{3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)pyridin-3-yl]phenyl}-2-oxo-5-oxazolidinyl]methyl]phosphate,
DA 7157
THERAPEUTIC CLAIM Treatment of complicated skin and skin structure infections
CHEMICAL NAMES
1. 2-Oxazolidinone, 3-[3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)-3-pyridinyl]phenyl]-5- [(phosphonooxy)methyl]-, (5R)-
2. [(5R)-3-{3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)pyridin-3-yl]phenyl}-2-oxooxazolidin-5- yl]methyl hydrogen phosphate
CHEMICAL NAMES
1. 2-Oxazolidinone, 3-[3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)-3-pyridinyl]phenyl]-5- [(phosphonooxy)methyl]-, (5R)-
2. [(5R)-3-{3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)pyridin-3-yl]phenyl}-2-oxooxazolidin-5- yl]methyl hydrogen phosphate
MOLECULAR FORMULA C17H16FN6O6P
MOLECULAR WEIGHT 450.3
TRADEMARK None as yet
SPONSOR Trius Therapeutics
CODE DESIGNATION TR-701 FA
CAS REGISTRY NUMBER 856867-55-5
Note: This adoption statement supersedes the USAN torezolid phosphate (N09/81), which is hereby rescinded and replaced by the USAN tedizolid phosphate (N10/118).\
TRADEMARK None as yet
SPONSOR Trius Therapeutics
CODE DESIGNATION TR-701 FA
CAS REGISTRY NUMBER 856867-55-5
Note: This adoption statement supersedes the USAN torezolid phosphate (N09/81), which is hereby rescinded and replaced by the USAN tedizolid phosphate (N10/118).\
...................................
Tedizolid, 856866-72-3
(5R)-3-{3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)pyridin-3-yl]phenyl}-5-(hydroxymethyl)-1,3-oxazolidin-2-one
(5R)-3-[3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)-3-pyridinyl]phenyl]-5-(hydroxymethyl)-2-oxazolidinone,
TR 700
- Molecular Formula: C17H15FN6O3
- Average mass: 370.337799
Torezolid (also known as TR-701 and now tedizolid[1]) is an oxazolidinone drug being developed by Trius Therapeutics (originator Dong-A Pharmaceuticals) for complicated skin and skin-structure infections (cSSSI), including those caused by Methicillin-resistantStaphylococcus aureus (MRSA).[2]
As of July 2012, tedizolid had completed one phase III trial, with another one under way. [3]Both trials compare a six-day regimen of tedizolid 200mg once-daily against a ten-day regimen of Zyvox (linezolid) 600mg twice-daily.
The prodrug of tedizolid is called “TR-701″, while the active ingredient is called “TR-700″.[4][5]
Trius Therapeutics will soon be reporting data from its second phase III trial (ESTABLILSH-2) and the recently announced publication of the data from its first phase III trial (ESTABLISH-1) in the Journal of the American Medical Association (JAMA)
- “Trius grows as lead antibiotic moves forward”. 31 Oct 2011.
- “Trius Completes Enrollment In Phase 2 Clinical Trial Evaluating Torezolid (TR-701) In Patients With Complicated Skin And Skin Structure Infections”. Jan 2009.
- http://clinicaltrials.gov/ct2/results?flds=Xf&flds=a&flds=b&term=tedizolid&phase=2&fund=2&show_flds=Y
- PMID 19528279 In vitro activity of TR-700, the active ingredient of the antibacterial prodrug TR-701, a novel oxazolidinone antibacterial agent.
- PMID 19218276 TR-700 in vitro activity against and resistance mutation frequencies among Gram-positive pathogens.
Emergence of bacterial resistance to known antibacterial agents is becoming a major challenge in treating bacterial infections. One way forward to treat bacterial infections, and especially those caused by resistant bacteria, is to develop newer antibacterial agents that can overcome the bacterial resistance. Coates et al. (Br. J. Pharmacol. 2007; 152(8), 1147-1154.) have reviewed novel approaches to developing new antibiotics. However, the development of new antibacterial agents is a challenging task. For example, Gwynn et al. (Annals of the New York Academy of Sciences, 2010, 1213: 5-19) have reviewed the challenges in the discovery of antibacterial agents.
Several antibacterial agents have been described in the prior art (for example, see PCT International Application Nos. PCT/US2010/060923, PCT/EP2010/067647, PCT/US2010/052109, PCT/US2010/048109, PCT/GB2009/050609, PCT/EP2009/056178 and PCT/US2009/041200). However, there remains a need for potent antibacterial agents for preventing and/or treating bacterial infections, including those caused by bacteria that are resistant to known antibacterial agents.
Various oxazolidinone-containing compounds have been disclosed for use asantibiotics. For example, oxazolidinone-containing compounds have been described in U.S. patent application Ser. No. 10/596,412 (filed Dec. 17, 2004), and WO 04/048350, WO 03/022824 and WO 01/94342, which are incorporated herein by reference.
U.S. patent application Ser. No. 12/577,089 (filed Oct. 9, 2009) and U.S. patent application Ser. No. 12/699,864 (filed Feb. 3, 2010), which are assigned to the same assignee as in the present application, disclose phosphate dimer impurities made during the process of making of the compounds disclosed therein. Surprisingly, it has been found that compounds containing at least two phosphates binding two oxazolidinone-containing moieties, such as dimers of oxazolidinone-containing compounds have antibacterial activity similar to their dihydrogen monophosphate analog
active drug of Formula I is (5R)-3-[3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)-3-pyridinyl]phenyl]-5-(hydroxymethyl)-2-oxazolidinone, i.e.,
These active compounds have been disclosed in WO 05/058886 and US Patent Publication No. 20070155798, while processes for making these and related compounds have been disclosed in U.S. patent application Ser. No. 12/577,089 (filed Oct. 9, 2009), and a crystalline form of the phosphate ester and related salts of the above compound has been disclosed in U.S. patent application Ser. No. 12/699,864 (filed Feb. 3, 2010).
US Patent Publication No. 20070155798, recently disclosed a series of potently anti-bacterial oxazolidinones including

wherein R═H, PO(OH)2, and PO(ONa)2.
Cubist Announces Submission of New Drug Application for Investigational Antibiotic Tedizolid for Treatment of Serious Skin Infections
LEXINGTON, Mass.–(BUSINESS WIRE)– Cubist Pharmaceuticals, Inc. today announced that it has submitted a New Drug Application (NDA) to the U.S. Food and Drug Administration (FDA) for approval of its investigational antibiotic tedizolid phosphate (TR-701). Cubist is seeking approval of tedizolid phosphate for the treatment of acute bacterial skin and skin structure infections (ABSSSI). Tedizolid phosphate is a once daily oxazolidinone being developed for both intravenous (I.V.) and oral administration for the treatment of serious Gram-positive infections, including those caused by methicillin-resistant Staphylococcus aureus (MRSA).
....................................................................
Espinoza-González NA, Welsh O, de Torres NW, Cavazos-Rocha N, Ocampo-Candiani J, Said-Fernandez S, Lozano-Garza G, Choi SH, Vera-Cabrera L.
Molecules. 2008 Jan 11;13(1):31-40.
Molecules. 2008 Jan 11;13(1):31-40.
..................................................
imp patents
| US2010305069 | 12-3-2010 | OXAZOLIDINONE CONTAINING DIMER COMPOUNDS, COMPOSITIONS AND METHODS TO MAKE AND USE |
| US7816379 | 10-20-2010 | Oxazolidinone derivatives |
| US2009192197 | 7-31-2009 | NOVEL OXAZOLIDINONE DERIVATIVES |
..................................................
TEDIZOLID disodium salt
59 nos in
38 nos
Tedizolid (formerly known as torezolid or TR-700) is the active hydroxymethyl oxazolidinone having the following formula:

Pharmaceutical prodrugs such as tedizolid phosphate (also referred to as TR-701, torezolid phosphate, and TR-701 “free acid” or FA) have the following formula:

The disodium salt of tedizolid phosphate, has the following structure:

..................................................................................................................................................................................
Example 1 Preparation of the Phosphate Monohydrogen Diester, Formula III
In this and the following Examples, “Formula III” refers to a compound wherein Z is
Figure US20100305069A1-20101202-C00024
and M=OH.
A 1-L, three-neck round-bottom flask equipped with a magnetic stirrer, nitrogen inlet/outlet and thermocouple was charged with the compound of Formula Ia below (16.0 g, 0.0499 mol], THF (320 mL, 20 vol) and Et3N (21.9 g, 0.216 mol, 5.0 equiv.).
Figure US20100305069A1-20101202-C00025
POCl3 (3.31 g, 0.0216 mol, 0.5 equiv.) was added dropwise via syringe over 5 minutes. The reaction temperature was maintained below 25° C. The batch was aged for 16 hours at room temperature at which point HPLC analysis (XBridge, C18) indicated that the reaction was complete. The reaction vessel was then immersed in an ice-water bath and a 500-mL addition funnel charged with 320 mL of H2O was attached to the reaction vessel. When the temperature of the reaction reached 2.7° C., H2O was added drop wise over 30 minutes. The temperature of the reaction was maintained below 10° C. Upon completion of the H2O addition, the ice-water bath was removed and the batch was aged for 3 hours. The solution was transferred to a 2-L round-bottom flask and concentrated under reduced pressure on a rotary evaporator. After removal of most of the THF from the solution, the aqueous mixture was extracted with 5 1-L portions of CH2Cl2:MeOH (9:1). The CH2Cl2 layers were combined and concentrated to a dark oil. This crude material was purified on 200 g of silica gel, eluting with 10% MeOH/CH2Cl2 to 20% 2 N NH3 in MeOH/CH2Cl2. Fractions containing mostly the bis-ester (as judged by TLC Rf=0.3 eluting with 20% 2 N NH3 in MeOH/CH2Cl2) were combined and concentrated under reduced pressure on a rotary evaporator, during which time a white precipitate was observed. The flask containing the slurry was removed from the rotary evaporator and equipped with a magnetic stir bar and allowed to stir while cooling to room temperature over 3 hours, during which time the slurry thickened. The solid was filtered and dried in a vacuum oven at 45° C. for 16 hours to give 3.55 g of bis-ester as an off-white solid (20% yield). HPLC analysis (Method A): 99.0% (AUC), tR=16.3 min. This reaction was repeated and the combined lots of the compound of Formula III (6.7 g) were slurried in 100 mL of MeOH (15 vol). The slurry was heated to 40° C. for 30 minutes and then allowed to cool to room temperature over 1 hour. The off-white solid was filtered and dried in a vacuum oven at 40° C. for 16 hours to give 6.15 g of the compound of Formula III (92% yield). The 1H NMR analysis of the product was consistent with the assigned structure. HPLC analysis (Method A): 99.0% (AUC), tR=16.3 min.
Example 2 Preparation of the Diphosphate Dihydrogen Diester, Formula IV
In Examples 2-5, “Formula IV” refers to a compound wherein Z is
Figure US20100305069A1-20101202-C00026
n=0 and M=O-imidazolium salt.
A 250-mL 3-neck round-bottom flask equipped with a magnetic stirrer, nitrogen inlet/outlet and thermocouple was charged with the compound of Formula IIa below (5.0 g, 11.1 mmol), carbonyldiimidazole (890 mg, 5.55 mmol, 0.5 equiv.) and DMF (100 mL, 20 vol).
Figure US20100305069A1-20101202-C00027
The suspension was heated to 50° C. and held at that temperature for 4 hours at which point HPLC analysis (XBridge, C18) indicated that the reaction was complete. The reaction was filtered at 50° C. and dried in a vacuum oven at 50° C. for 24 hours to give 5.15 g of the imidazolium salt (i.e., the compound of Formula IV) as an off-white solid (98% yield). The 1H NMR analysis of the product was consistent with the assigned structure. HPLC analysis (Method A): 94.5% (AUC), tR=14.6 min.
TABLE 1
Method A (Waters XBridge C18 Column)
Time (min) Flow (mL/min) % A % B
0.0 1.0 98.0 2.0
15.0 1.0 5.0 95.0
25.0 1.0 5.0 95.0
27.0 1.0 98.0 2.0
30.0 1.0 98.0 2.0
A = 87% 25 mM ammonium bicarbonate solution in water/13% Acetonitrile
B = Acetonitrile
Wavelength = 300 nm
 disodium salt is TR 701
disodium salt is TR 701
..........................................
US8580767
Various oxazolidinone-containing compounds have been disclosed for use as antibiotics. For example, oxazolidinone-containing compounds have been described in U.S. patent application Ser. No. 10/596,412 (filed Dec. 17, 2004), and WO 04/048350, WO 03/022824 and WO 01/94342, which are incorporated herein by reference.
U.S. patent application Ser. No. 12/577,089 (filed Oct. 9, 2009) and U.S. patent application Ser. No. 12/699,864 (filed Feb. 3, 2010), which are assigned to the same assignee as in the present application, disclose phosphate dimer impurities made during the process of making of the compounds disclosed therein. Surprisingly, it has been found that compounds containing at least two phosphates binding two oxazolidinone-containing moieties, such as dimers of oxazolidinone-containing compounds have antibacterial activity similar to their dihydrogen monophosphate analog,
These active compounds have been disclosed in WO 05/058886 and US Patent Publication No. 20070155798, while processes for making these and related compounds have been disclosed in U.S. patent application Ser. No. 12/577,089 (filed Oct. 9, 2009), and a crystalline form of the phosphate ester and related salts of the above compound has been disclosed in U.S. patent application Ser. No. 12/699,864 (filed Feb. 3, 2010). The latter two applications are assigned to the same assignee as in the present application
.........................................................................................................................................
SYNTHESIS
US20070155798

DESCRIPTION OF COMPDS
10,
(R)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-5-hydroxymethyl oxazolidin-2-on (compound 10)

...........................................................................................................................................
18
Preparation of (R)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-5-fluoromethyl oxazolidin-2-on (compound 18)
...............................................................................................................................................................................
33
(R)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-5-methoxymethyl oxazolidin-2-on (compound 33)
..............................................................................................................................................................
59
(R)-[3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-2-oxo-5-oxazolidinyl]methyl disodiumphosphate (compound 59)

...........................................................................................................................................................................
72
mono-[(R)-[3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-2-oxo-5-oxazolidinyl]methyl]phosphate (compound 72)

COMPLETE SYNTHESIS
Example 5
Preparation of 2-cyano-5-bromopyridine
In 1 L of dimethylformamide was dissolved 100 g of 2,5-dibromopyridine, 32 g of cupper cyanide and 17.8 g of sodium cyanide were added to the solution at room temperature and the solution was stirred at the temperature of 150° C. for 7 hours for reaction. After being cooled to room temperature, the reaction mixture was added with water and extracted with ethyl acetate. The organic layer was washed with brine, dehydrated, filtered and concentrated in vacuo. The title compound 54 g was obtained. Yield 70%.
1HNMR(CDCl3) δ 8.76(s,1H), 7.98(dd,1H), 7.58(dd,1H)
Example 6
Preparation of 2-(tetrazol-5-yl)-5-bromopyridine
10 g of 2-cyano-5-bromopyridine prepared in the Preparation example 5 was dissolved in 100 ml of dimethylformamide, 5.33 g of sodiumazide, and 4.4 g of ammonium chloride were added to the solution at room temperature, and the solution was stirred at the temperature of 110° C. for 3 hours for reaction. The reaction mixture was added with water and then was extracted with ethyl acetate. The organic layer, thus separated, was washed with brine, dehydrated, filtrated and concentrated in vacuo thereby to obtain 10.5 g of the title compound. Yield 85%.
Preparation Example 7 Preparation of 2-(1-methyltetrazol-5-yl)-5-bromopyridine and 2-(2-methyltetrazol-5-yl)-5-bromopyridine
10.5 g of 2-(tetrazol-5-yl)-5-bromopyridine prepared in the Preparation example 6 was dissolved in 100 ml of dimethylformamide. And then 6.5 g of sodium hydroxide was added to the solution and 9.3 g of iodomethane was slowly added to the solution at the temperature of 0° C. The solution was stirred for 6 hours at room temperature, added with water, extracted with ethyl acetate. And then the organic layer was washed with brine, dehydrated, filtrated, concentrated in vacuo and purified by column chromatography to obtain 4 g of 2-(1-methyltetrazol-5-yl)-5-bromopyridine and 5 g of 2-(2-methyltetrazol-5-yl)-5-bromopyridine.
1) 2-(1-methyltetrazol-5-yl)-5-bromopyridine
1HNMR(CDCl3) δ 8.77(t,1H), 8.23(dd,1H), 8.04(dd,1H), 4.46(s,3H)
2) 2-(2-methyltetrazol-5-yl)-5-bromopyridine
1HNMR(CDCl3) δ 8.80(t,1H), 8.13(dd,1H), 7.98(dd,1H), 4.42(s,3H)
Example 1
Preparation of N-Carbobenzyloxy-3-fluoroaniline
3-fluoroaniline 100 g was dissolved in 1 L of tetrahydrofuran (THF) and the solution was added with 150 g (1.8 mol) of sodium bicarbonate (NaHCO3). After being cooled to 0° C., the solution was slowly added with 154 ml of N-carbobenzyloxy chloride (CbzCl) for reaction. While the temperature was maintained at 0° C., the reaction mixture was let to react for 2 hours with stirring. Afterwards, the reaction was extracted with 0.5 L of ethyl acetate. The organic layer, after being separated, was washed with brine, dried over anhydrous magnesium sulfate (MgSO4) and concentrated in vacuo. The residue was washed twice with n-hexane to afford the title compound as white crystal. 132 g. Yield 85%.
Example 2
Preparation of (R)-3-(3-fluorophenyl)-2-oxo-5-oxazolidinylmethanol
132 g of N-carbobenzyloxy-3-fluoroaniline 132 g prepared in the Preparation example 1 was dissolved in 1.3 L of tetrahydrofuran and the solution was cooled to −78° C. 370 ml of n-buthyllitium (n-BuLi, 1.6M/n-hexane) was slowly added to the solution in a nitrogen atmosphere, followed by stirring for 10 min. And 84 ml of (R)-(−)-glycidylbuthylate was slowly added to the reaction mixture, stirred at the same temperature for 2 hours and allowed to react for 24 hours at room temperature. After completion of the reaction, the solution was added with ammonium chloride (HH4Cl) solution and extracted with 0.5 L of ethyl acetate at room temperature. The organic layer, thus separated, was washed with brine, dried over anhydrous magnesium sulfate and concentrated in vacuo. The residue was dissolved in 100 ml of ethyl acetate and washed with n-hexane to give white crystals, which were purified to the title compound. 80 g. Yield 70%.
1H NMR (DMSO-d6) δ 7.85(t,1H), 7.58(dd,1H), 7.23(dd,1H), 4.69(m,1H), 4.02 (t,1H), 3.80(dd,1H), 3.60(br dd,2H).
Example 3
Preparation of (R)-3-(4-iodo-3-fluorophenyl)-2-oxo-5-oxazolidinylmethanol
In 300 ml of acetonitryl was dissolved 30 g of (R)-3-(3-fluorophenyl)-2-oxo-5-oxazolidinylmethanol prepared in the Preparation example 2, and 46 g of trifluoroacetic acid silver salt (CF3COOAg) and 43 g of iodide were added to the solution. After being stirred for one day at room temperature, the solution was added with water and was extracted with ethyl acetate. The organic layer, thus separated, was washed with brine and dehydrated. And then the residue was filtered, concentrated in vacuo and dried thereby to form the title compound 44 g. Yield 94%.
1H NMR (DMSO-d6) δ 7.77(t,1H), 7.56(dd,1H), 7.20(dd,1H), 5.20(m,1H), 4.70 (m,1H), 4.07(t,1H), 3.80(m,1H), 3.67(m,2H), 3.56(m,3H)
Example 4
Preparation of (R)-3-(4-tributhylstannyl-3-fluorophenyl)-2-oxo-5-oxazolidinylmethanol
In 660 ml of 1,4-dioxan was dissolved 50 g of (R)-3-(4-iodo-3-fluorophenyl)-2-oxo-5-oxazolidinylmethanol prepared in the Preparation example 3, 52 g of hexabutylditin ((Bu3Sn)2) and 9.3 g of dichlorobistriphenylphosphinpalladium were added into the solution, and stirred for 2 hours. The solution was filtered using celite and concentrated in vacuo. The residue was purified by column chromatography and 45 g of the title compound was formed.
1H NMR (DMSO-d6) δ 7.74(m,3H), 5.20(t,1H), 4.71(m,1H), 4.08(t,1H), 3.82(dd,1H), 3.68(m,1H), 3.52(m,1H), 1.48(m, 6H), 1.24(m, 6H), 1.06(m,6H), 0.83(t,9H)
COMPD 10
Example 1 Preparation of (R)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-5-hydroxymethyl oxazolidin-2-on (compound 10)
In 150 ml of 1-methyl-2-pyrrolidone was dissolved 37 g of (R)-3-(4-tributhylstannyl-3-fluorophenyl)-2-oxo-5-oxazolidinylmethanol. The solution was added with 19.7 g of 2-(2-methyltetrazol-5-yl)-5-bromopyridine, 10.44 g of lithium chloride and 2.9 g of dichlorobistriphenylphospine palladium(II) at room temperature and then stirred at the temperature of 120° C. for 4 hours. The reaction mixture was added with water and then extracted with ethyl acetate. The organic layer, thus separated, was washed with brine, dehydrated, filtrated, concentrated in vacuo and purified by column chromatography to provide 8 g of the title compound. Yield 26%.
1H NMR (DMSO-d6) δ 8.90(s,1H), 8.18(m,2H), 7.70(m,2H), 7.49(dd,1H), 5.25(t,1H), 4.74(m,1H), 4.46(s,3H), 4.14(t,1H), 3.88(dd,1H), 3.68(m,1H), 3.58 (m,1H)
COMPD 18
Example 28 Preparation of (R)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-5-fluoromethyl oxazolidin-2-on (compound 18)
In 5 ml of methylenchloride was dissolved 100 mg of the compound 10. The solution was added with 43 mg of diethylaminosulfurtrifloride (DAST) and 0.078 ml of triethylamine and then stirred for 24 hours. After being concentrating, the reaction mixture was purified by column chromatography to obtain the title compound 75 mg. Yield 75%.
1H NMR (DMSO-d6) δ 8.91(s,1H), 8.19(m,2H), 7.74(t,1H), 7.66(dd,1H) 7.49 (dd,1H), 5.06(m,1H), 4.89(m,2H), 4.46(s,3H), 4.23(t,1H), 3.95(dd,1H)
COMPD 33
Example 37 Preparation of (R)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-5-methoxymethyl oxazolidin-2-on (compound 33)
In 10 ml of methanol was dissolved 400 mg of (R)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-5-methansulfonyloxymethyl oxazolidin-2-on prepared in the secondary step of the Example 24. The solution was added with 90 mg of sodium methoxide at room temperature and then stirred for one day at room temperature. The solution was extracted with ethyl acetate and the organic layer, thus separated, was washed with water and brine. The organic layer was dehydrated, filtered, concentrated in vacuo and purified by column chromatography to provide the title compound 200 mg. Yield 58%.
1H NMR(CDCl3) δ 8.90(s,1H), 8.29(d,1H), 8.04(d,1H), 7.61(dd,1H), 7.58 (t,1H), 7.38(dd,1H), 4.80(m,1H), 4.45(s,3H), 4.08(t,1H), 3.96(dd,1H), 3.67 (m,2H), 3.43(s,3H)
COMPD 59
Example 58 Preparation of mono-[(R)-[3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-2-oxo-5-oxazolidinyl]methyl]phosphate (compound 72) and (R)-[3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-2-oxo-5-oxazolidinyl]methyl disodiumphosphate (compound 59)
1. The Primary Step
In 10 ml of mixture solvent (tetrahydrofuran:methylenchloride=1:1) was dissolved 1 g of compound 10. The solution was added with 0.6 g of tetrazole and 2.3 g of di-tetrabutyl diisoprophylphosphoamidite and stirred for 15 hours at room temperature. The reaction mixture was refrigerated to −78° C., added with 0.7 g of metachloroperbenzoic acid and stirred for 2 hours. After being cooling to −78° C., the reaction mixture was added with metachloroperbenzoic acid (0.7 g). When the reaction mixture was stirred for 2 hours, the temperature of the reaction mixture was raised to room temperature. The reaction mixture was then added with ethyl acetate. The organic layer, thus separated, was washed with sodium bisulfate, sodium bicarbonate and brine, dehydrated, filtered and concentrated in vacuo, followed by purification with column chromatography thereby to provide (R)-[3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-2-oxo-5-oxazolidinyl]methyl phosphoric acid ditetrabuthylester (0.71 g, 71%).
1H NMR (DMSO-d6) δ 8.90(s,1H), 8.18(m,2H), 7.74(t,1H), 7.68 (dd,1H), 7.49(dd,1H), 4.98(m,1H), 4.46(s,3H), 4.23(t,1H), 4.18(m,1H), 4.09(m,1H), 3.89 (dd,1H), 1.39(s,9H), 1.38(s,9H)
The crystal prepared the above method was dissolved in a mixture of methanol and chloroform. And then the solution added with 3.4 ml of sodium methoxide (0.3M methanol solution) at the room temperature and stirred for 10 hours. The reaction mixture was concentrated to prepare the residue. The residue was crystallized and filtered thereby to obtain the title compound (compound 59) 300 mg.
1H NMR (D2O) δ 8.27(s,1H), 7.56(dd,2H), 7.06(m,2H), 6.90(m,1H), 4.79 (m,1H), 4.63(s,3H), 3.90(m,4H)
COMPD 72
The Secondary Step
In 30 ml of methylenchloride was dissolved the compound (0.7 g) in the Primary Step. The solution was added with 15 ml of trifluoroacetic acid and then stirred for 1 hour at room temperature. The reaction mixture was concentrated in vacuo to prepare the residue. The residue was crystallized with ethanol and ethyl ether to obtain mono-[(R)-[3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-2-oxo-5-oxazolidinyl]methyl]phosphate (compound 72) 400 mg.
1H NMR (DMSO-d6) δ 8.92(s,1H), 8.20(m,2H), 7.74(t,1H), 7.66(dd,1H), 7.500(dd,1H), 4.95 (m,1H), 4.46(s,3H), 4.21(t,1H), 4.05(m,2H), 3.91(dd,1H)
US20070155798
...............................................................
IMPURITIES
US8426389
.....................................................
2 RADEZOLID


























4.....................




5.........................
EPEREZOLID













.....................................
6.........................
MRX 1
 MRX I
MRX I








.................................
7...................
LCB01-0371







...............................................................
8 RANBEZOLID










Substitution of five-membered heterocycles on to the ‘piperazinyl–phenyl–oxazolidinone’ core structure led to the identification of ranbezolid as a clinical candidate. Further replacement of piperazine ring with other diamino-heterocycles led to compounds with potent antibacterial activity.


...................................
9 FYL 67






10
WCK ?........http://newdrugapprovals.org/2015/11/24/new-antibacterial-oxazolidinones-in-pipeline-by-wockhardt/

WCK ?
(5S)-N-{3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide
(5S)-N- {3-[3,5-difluoro-4-(4-hydroxy-(4-methoxymethyl)-piperidin- lyl)phenyl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide
MF C19 H25 F2 N3 O5, MW 413.42
Acetamide, N-[[(5S)-3-[3,5-difluoro-4-[4-hydroxy-4-(methoxymethyl)-1-piperidinyl]phenyl]-2-oxo-5-oxazolidinyl]methyl]-
CAS 957796-51-9
Antibacterial oxazolidinones
Wockhardt Ltd, Innovator
Wockhardt Research Center,
WO 2015173664, US8217058, WO 2012059823, IN 2011MU03726

Oxazolidinone represent a novel chemical class of synthetic antimicrobial agents. Linezolid represents the first member of this class to be used clinically. Oxazolidinones display activity against important Gram-positive human and veterinary pathogens including Methicillin-Resistant Staphylococcus aureus (MRSA), Vancomycin Resistant Enterococci (VRE) and β-lactam Resistant Streptococcus pneumoniae (PRSP). The oxazolidinones also show activity against Gram-negative aerobic bacteria, Gram-positive and Gram-negative anaerobes. (Diekema D J et al., Lancet 2001 ; 358: 1975-82).
Various oxazolidinones and their methods of preparation are disclosed in the literature. International Publication No. WO 1995/25106 discloses substituted piperidino phenyloxazolidinones and International Publication No. WO 1996/13502 discloses phenyloxazolidinones having a multisubstituted azetidinyl or pyrrolidinyl moiety. US Patent Publication No. 2004/0063954, International Publication Nos. WO 2004/007489 and WO 2004/007488 disclose piperidinyl phenyl oxazolidinones for antimicrobial use.
Pyrrolidinyl/piperidinyl phenyl oxazohdinone antibacterial agents are also described in Kim H Y et al., Bioorg. & Med. Chem. Lett., (2003), 13:2227-2230. International Publication No. WO 1996/35691 discloses spirocyclic and bicyclic diazinyl and carbazinyl oxazolidinone derivatives. Diazepeno phenyloxazolidinone derivatives are disclosed in the International Publication No. WO 1999/24428. International Publication No. WO 2002/06278 discloses substituted aminopiperidino phenyloxazolidinone derivatives.
Various other methods of preparation of oxazolidinones are reported in US Patent No. 7087784, US Patent No. 6740754, US Patent No. 4948801 , US Patent No. 3654298, US Patent No. 5837870, Canadian Patent No. 681830, J. Med. Chem., 32, 1673 (1989), Tetrahedron, 45, 1323 (1989), J. Med. Chem., 33, 2569 (1990), Tetrahedron Letters, 37, 7937-40 (1996) and Organic Process Research and Development, 11 , 739-741(2007).
Indian Patent Application No. 2534/MUM/2007 discloses a process for the preparation of substituted piperidino phenyloxazolidinones. International Publication No. WO2012/059823 further discloses the process for the preparation of phosphoric acid mono-(L-{4-[(5)-5-(acetylaminomethyl)-2-oxo-oxazolidin-3-yl]-2,6-difluorophenyl}4-methoxymethyl piperidine-4-yl)ester.
US Patent No. 8217058 discloses (5S)-N-{3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-l-yl)-phenyl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide as an antibacterial agent and its process for preparation.
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015173664&recNum=1&maxRec=&office=&prevFilter=&sortOption=&queryString=&tab=PCTDescription

In some embodiments, there is provided a process for preparation of a compound of Formula (I) as shown in Scheme 1

(I I) (I N)
Scheme 1

Example 1
Preparation of (55)-iV-{3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-l-yl)- phenyl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide (I)
To a stirred solution of lithium teri-butoxide (59.1 g, 0.74 mol) in tetrahydrofuran (500 ml) was added a solution of [3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-l-yl)-phenyl]-carbamic acid benzyl ester (II) (100 g, 0.25 mol) in 500 ml of tetrahydrofuran slowly at room temperature. The resulting mixture was stirred for 3 hours at room temperature (formation of lumps observed). The reaction mixture was cooled to temperature of 10°C to 15°C and acetic acid l-(acetylamino-methyl)-2-chloro-ethyl ester (III) (95.2 g, 0.49 mol) was added in one lot, after 5 minutes methanol (2.36 g, 0.075 mol) was added in one portion. The resulting mixture was stirred further at temperature of 10°C to 15°C. After 5 hours the reaction mixture was allowed to warm to room temperature and stirring continued further for 16 hours. An aqueous solution of saturated ammonium chloride (100 ml) was added to the reaction mixture, the resulting mixture was stirred well and the solvent evaporated under reduced pressure (35°C, 150 mm Hg). The residual mixture was diluted with water (1 L stirred well and filtered under suction, the residual solid was washed with additional fresh water (100 ml). The residual mass was suspended in acetone (500 ml), stirred well and the mixture diluted with hexane (1 L), slowly. The mixture was stirred further for 1 hour and filtered under suction. The residual solid was washed with a 2:1 mixture of acetone and water (100 ml). The residual solid was dried at 45°C, for 3.5 hour at 4 mm Hg, to obtain the 78 g of (55)-N-{3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-l -yl)-phenyl]-2-oxo-oxazolidin-5-ylmethylj -acetamide (I) as white solid, in 77% yield.
Analysis:
Mass: 414 (M+l ); for Molecular Weight: 413 and Molecular Formula:
Melting Point: 178-179°C;
1H NMR (400 MHz, DMSO): δ 8.18-8.21 (m, 1H), 7.19-7.25 (d, 2H), 4.07-4.71 (m, 1H), 4.32 (s, 1H), 4.02-4.07 (t, 1H), 3.64-3.68 (t, 1H), 3.14 (s, 2H), 2.81-2.83 (d, 2H), 1.81 (s, 3H), 1.63-1.69 (t, 2H), 1.42-1.45 (d, 2H);
Purity as determined by HPLC: 97.65%.
Example 2
Preparation of acetic acid l-(acetylamino-methyl)-2-chloro-ethyl ester (III)
Step-I: Preparation of l-amino-3-chloro-propan-2-ol hydrochloride (VI)
Benzaldehyde (118.67 g, 1.03 mol) was dissolved in ethanol (297 ml) under stirring and the solution was cooled to 18-19°C. To this solution aqueous ammonia solution (25%) (101.58 ml) was added slowly, followed by slow addition of S-epichlorohydrin (100 g, 1 mol). The resulting mixture was warmed to 40°C and stirred for 7 hours. The mixture was allowed to cool to room temperature and stirred further. After 16 hours, the reaction mixture was concentrated to 50% volume under reduced pressure. Toluene (228 ml) was added to the reaction mixture followed by addition of aqueous hydrochloric acid (162 ml of concentrated hydrochloric acid diluted with 152 ml of water). The mixture thus obtained for 3 hours at 45°C, the resulting mixture was allowed to cool to room temperature and the toluene layer separated. The toluene layer was further extracted with water (56 ml). The combined aqueous layer was diluted with ethanol (56 ml) and the mixture evaporated under reduced pressure. This process was repeated again. To the final concentrate was added ethanol (180 ml), stirred for 10 minutes and the mixture cooled to -28°C to -30°C and maintained at this temperature for 2 hours. The separated solid was filtered under suction and the residue washed with cold (-30°C) ethanol (50 ml). The residue was dried at 45°C, under reduced pressure (4 mm Hg) for 3 hours, to obtain 96 g of l-amino-3-chloro-propan-2-ol hydrochloride (VI) as white solid in 61% yield.
Analysis:
Mass: 110 (M+l) as free base; for Molecular Weight: 145.5 and Molecular Formula:
1H NMR (400 MHz, D20): δ 4.02-4.08 (m, 1H), 3.51-3.61 (m, 2H), 3.12-3.16 (dd, 1H), 2.93 -2.99 (dd, 1H).
Step-II: Preparation of acetic acid l-(acetylamino-methyl)-2-chloro-ethyl ester (III).
A stirred solution of dichloromethane (220.8 ml) containing the step-I salt (96 g, 0.66 mol) was cooled to 18-20°C. Acetic anhydride (154.78 g, 1.5175 mol) was added slowly (slight exothermic). Pyridine (67.76 g, 0.8577 mol) was added slowly (exothermic) while maintaining the temperature at 18-20°C. The resulting mixture was heated to 40°C for 5 hours. The reaction mixture was allowed to cool to room temperature and stirring continued for further 16 hours. The reaction mass was cooled to 3-6°C and diluted with 170 ml of fresh water. To this was added an aqueous solution of potassium carbonate (191.2 g of K2CO3 in 382 ml water). The reaction mixture was further diluted with additional dichloromethane (170 ml) and water (425 ml). The reaction mass was stirred well and the dichloromethane layer separated. The aqueous layer was further extracted with 2×170 ml dichloromethane. The combined dichloromethane layer was washed with aqueous sodium chloride solution (13.6 g of sodium chloride in 493 ml water). The solvent was evaporated till a volume of 170 ml and the residual layer was diluted with toluene (340 ml), stirred well and the solvent was evaporated completely at 40°C under reduced pressure (4 mm Hg). To the residue ethyl acetate (170 ml) and hexane (187 ml) were added and the mixture stirred for 30 minute. The separated solid was filtered under suction and the residue washed with 50 ml of a 1 :1 mixture of ethyl acetate and hexane. The solid obtained was dried under reduced pressure (4 mm Hg) at 45°C for 3.5 hours, to obtain 96 g of acetic acid l-(acetylamino-methyl)-2-chloro-ethyl ester (III) as a white solid, in 75% yield.
Analysis:
Mass: 194 (M+l); for Molecular Weight: 193 and Molecular Formula: C7Hi2ClN03; 1H NMR (400 MHz, CDC13): 5 5.69 (s, 1H), 5.0-5.1 (m, 1H), 3.4-3.7 (m, 4H), 2.1 (s, 3H), 1.9 (s, 3H).

Wockhardt Ltd,

 (3) (4)
(3) (4)
Scheme -1
 (6) Formula π Scheme-2
(6) Formula π Scheme-2
 Formula II Formula in
Formula II Formula in
 Formula I(a) Scheme-4
Formula I(a) Scheme-4
Example -11 : (5S)-N- {3-[3,5-difluoro-4-(4-hydroxy-(4-methoxymethyl)-piperidin- lyl)phenyl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide
The example- 10 (54.86 g, 0.144 mol) was suspended in methanol (1100 ml) under stirring at RT. Sodium metal (4 g, 0.174 mol) was added in small lots in 2 min to the above suspension under stirring. The reaction mixture was warmed to 40-420C and was stirred at this temperature for about 40 hrs. After completion of the reaction (TLC), the solvent was evaporated under reduced pressure to obtain a thick slurry. The thick slurry thus obtained was gradually added to water (1100 ml) under stirring. After the complete addition, the pH of the aqueous suspension was adjusted to 7 by adding sufficient quantity of glacial acetic acid. The separated solid was filtered and the residue was washed with water. The obtained solid was further purified by column chromatography over silica gel to obtain the product as a white solid, 32.7 g, 55 % yield.
M.P.: 173-1740C;
MS : M+l= 414(MH+, 100%); for M.F.: Ci9H25F2N3O5
1H-NMR (400 MHz, CDCl3): δ 7.0-7.1 (m, 2H5Ar-H), 6.0 (t, IH, NH), 4.70-4.80 (m, IH), 4.00 (t,lH), 3.70-3.75 (m, 2H), 3.5-3.7 (m, IH), 3.43 (s, 3H, OCH3), 3.37-3.42 (m, 2H), 3.30 (s, 2H, -OCH2), 3.0-3.05 (m, 2H), 2.22(bs,lH ,-OH),2.04 (s, 3H, COCH3), 1.70-1.75 (m, 4H).
Phosphoric acid mono-(1-{4-[(S)-5-(acetylamino-methyl)-2-oxo-oxazolidin-3-yl]-2,6-difluorophenyl}-4-methoxy methyl-piperidin-4-yl) ester

Specific intermediate compounds of the invention include:
6-(2,6-difluoro-4-nitrophenyl)-1-oxa-6-azaspiro[2.5]octane;
1-(2,6-Difluoro-4-nitro-phenyl)-4-methoxymethyl-piperidin-4-ol;
[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-carbamic acid benzyl ester;
(5R)-3-[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-5-hydroxymethyl-oxazolidin-2-one;
(5R)-Methanesulfonic acid 3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-2-oxo-oxazolidin-5-ylmethyl ester;
(5R)-3-[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-5-azidomethyl-oxazolidin-2-one; and
(5S)- N-{3-[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide.
Examples
Preparation of Intermediate-1: 1-(2,6-Difluoro-4-nitrophenyl)-piperidin-4-one
Chloroform (9.3 L) was charged in a 20 L reaction assembly and 4-piperidone hydrochloride (1.17 Kg, 7.62 mol) was added under stirring followed by triethylamine (2.14 Kg, 2.95 L, 21.1 mol). After 30 minutes of stirring, 3,4,5-trifluoronitrobenzene (1.5 Kg, 8.47 mol) was added to the mixture in one lot and the contents were heated to 65-70ºC for 8 h. After completion of the reaction, chloroform was removed under vacuum to obtain a syrupy mass. At this stage, water (10 L) was added to the mass and the chloroform recovery was continued under vacuum below 65oC till the chloroform was removed completely. The slurry was cooled to RT and filtered. The solid product was washed with water (3 L) followed by hexanes (2 L). The product was dried in a vacuum oven below 70oC to obtain the product as a yellow solid, 1.88 Kg ; Yield 97%.
M.P.: 130-132oC; MS: 257(M+1); M.F.: C11H10F2N2O3.
Preparation of Intermediate 3: 1-(2,6-Difluoro-4-nitro-phenyl)-4-methoxymethyl-piperidin-4-ol
Method A:
Preparation of Intermediate–2: (Stage-I): 6-(2,6-difluoro-4-nitrophenyl)-1-oxa-6-azaspiro[2.5]octane
A solution of trimethylsulfoxonium iodide (1.504kg, 6.836mol) in acetonitrile (7L) was cooled to 0 to 5oC. , under argon atmosphere. Potassium tert-butoxide (0.736kg, 6.552 mol) was added in small lots over 0.5h. The resulting solution was stirred for 2h at the same temperature. To this solution was added 1-(2,6-Difluoro-4-nitrophenyl)-piperidin-4-one ( 1.4kg, 5.46mol) in small lots over a period of 1h, while maintaining the temp. between 5-10oC. The resulting mixture was stirred for 1h. The solvent was evaporated to a minimum amount possible, under reduced pressure while maintaining the temperature below 10oC. The residue was poured in water( 18L) and the pH adjusted to neutral with dilute acetic acid. The resulting slurry was stirred well and the separated solid filtered under suction. The solid was washed with fresh water till the filtrate was free of acetic acid. The solid was dried at 80oC, for 6h, under reduced pressure to obtain the product as pale yellow solid, 1.264kgs, yield 85%.
M.P.: 96-97oC; MS: M+1: 271; M.F.: C12H12F2N2O3,.
Preparation of Intermediate-3: (Stage-II): 1-(2,6-Difluoro-4-nitro-phenyl)-4-methoxymethyl-piperidin-4-ol
To a solution of sodium methoxide (236g, 4.35mol) in methanol (3L), at RT, was added 6-(2,6-difluoro-4-nitrophenyl)-1-oxa-6-azaspiro [2.5]octane (964g, 3.57mol) in small portions and the reaction mixture was stirred for 26h at RT. Acetic acid (265g, 4.44mol) was added slowly to neutralize the pH of the solution. The resulting mixture was poured into chilled water(18L) and stirred for 1h. The separated solid was filtered under suction. The solid was washed with additional water till the filtrate was free of acetic acid. The solid was dried for 10hat RT under reduced pressure, to obtain the product as a pale yellow solid, 973g, yield, 90%
M.P.: 84-86oC; MS: 303 (M+1); M.F.: C13H16F2N2O4
Method B:
Dimethylsulfoxide (DMSO, 100 ml) and methanol (500 ml) were charged in a 1 L glass reaction assembly. Potassium hydroxide (59.2g, 0.898 mol) was charged in the assembly followed by trimethylsulfoxonium iodide (94.5 g, 0.43 mol) and the contents were stirred for 30 minutes and then cooled to 10oC-15oC. To the cooled contents was added 1-(2,6-difluoro-4-nitrophenyl)-piperidin-4-one (100 g, 0.39 mol) in small lots. After the addition, the temperature was allowed to raise to RT and the contents were further stirred for 24 h (ring opening of the epoxide intermediate viz. 6-(2,6-difluoro-4-nitrophenyl)-1-oxa-6-azaspiro[2.5]octane takes place).
[Physical data of the intermediate: M.P.: 96-970C, MS: 271(M+1); M.F.: C12H12F2N2O3, .
After completion of the reaction the contents were poured slowly in ice-water (600g crushed ice in 600 ml water). The precipitated solid product was filtered and was washed with water:methanol, 2:1 (100 ml X 2). The wet product was used in the next step.
M.P.: 84-86oC; MS: 303 (M+1);.M.F.: C13H16F2N2O4,:
Preparation of Intermediate -5: [3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-carbamic acid benzyl ester
Method A: Preparation of Intermediate 4: ( Stage-I)
Water (1.19 L) and methanol (595 ml) were charged in a 3 L glass reaction assembly, followed by 1-(2,6-difluoro-4-nitro-phenyl)-4-methoxymethyl-piperidin-4-ol (85 g, 0.281 mol) and the contents were stirred. Sodium dithionite (288 g, 1.407 mol) was added in one lot and the reaction mixture was heated to 80oC for 8 h. After completion of the reaction (TLC), methanol was recovered under vacuum below 65oC. After the recovery, the aqueous residue was extracted with chloroform (400 ml X 3). The combined chloroform extract (containing the intermediate 1-(4-amino-2,6-difluoro-phenyl)-4-methoxymethyl-piperidin-4-ol) was dried over anhydrous Sodium sulfate and used in the next step (carbamate formation).
Preparation of Intermediate -5: (Stage-II):
The above chloroform extract was charged in a 3 L glass reaction assembly. Sodium bicarbonate (70 g, 0.843 mol) was added to the extract and the contents were cooled to 15oC-20oC. Benzylchloroformate solution (50% in toluene, 48 g, 96 ml, 0.281 mol) was added slowly to the above mixture under stirring. After completion of the addition, the reaction mixture was stirred at RT for 2 h. After completion of the reaction (TLC), the contents were filtered on a Buchner assembly and the solid cake was washed with chloroform (85 ml X 2). The combined filtrate was evaporated under vacuum below 50oC to obtain yellowish oily mass, which was poured slowly in hexanes (850 ml) under stirring to obtain a precipitate. The precipitated product was filtered and washed with hexanes (100 ml X 2). The product was dried in a vacuum oven below 65oC to obtain 60.2 g brownish product (Yield = 38% on the basis of step-I input).
M.P.: 138-140oC; MS: 407(M+1); M.F.: C21H24F2N2O4.:.
Method B: : Preparation of Intermediate 4: ( Stage-I): To a solution of 1-(2,6-difluoro-4-nitro-phenyl)-4-methoxymethyl-piperidin-4-ol (973g, 3.22 mol) in ethyl acetae (10L) was added 10% Pd-C, (250g, 50% wet) and the resulting miture was hydrogenated in a pressure at 30 PSI, 45-55oC, for 3h. The catakyst was filtered and the residue was washed with additional ethyl acetate( 200ml). The combined filtrates were used as such for the next reaction (carbamate formation)
Preparation of Intermediate -5: (Stage-II):
To the above filtrate was added sodium bicarbonate(406g, 4.83 mol) and the mixture warmed to 40-45oC. To this mixture was added a 50% solution of Benzyl chloroformate in toluene(1.373L, 4.025 mol), drop-wise, over a period of 1h. Stir the resulting mixture for 1h and filter the insoluble material. The residue was washed with 300ml of ethyl acetate. The filtrates were combined and the solvent evaporated under reduced pressure, below 55oC.. Cool the residue and dilute it with hexane(10L). The resulting slurry was stirred well and the separated solid was filtered under suction. The residue was washed with additional hexane ( 2L). The solid was dried for 10h at RT, to obtain the product as dark brown solid, 1200g, yield, 96%.
M.P.: 138-140oC; MS: 407( M+1); M.F.: C21H24F2N2O.
Preparation of Intermediate -6:
(5R)-3-[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-5-hydroxymethyl-oxazolidin-2-one
To a mixture of [3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-carbamic acid benzyl ester (100g, 0.237 mol) in dry tetrahydrofuran (THF) (2 L) at 40ºC was added drop-wise n-BuLi in hexane (1.6M, 45.5 g, 455 ml, 0.711 mol) under nitrogen atmosphere. The contents were stirred for 1 h at 40ºC and R-(-)-glycidyl butyrate (68.25 g, 0.474 mol) was added gradually. After the addition of R-(-)-glycidyl butyrate, the reaction mixture was stirred for 5-6 h at 40oC till completion of the reaction (TLC). After completion of the reaction, a solution of sodium methoxide (2 g) in methanol (66 ml) was added to the contents followed by water (8 ml) and the contents were stirred for an additional 0.5 h. Water (1 L) was added to the solution and the contents were extracted with ethyl acetate (1 L). The aqueous layer was further extracted with ethyl acetate (3 X 500 ml). The combined organic layer was evaporated under vacuum to obtain a thick residue. tert-Butyl methyl ether (1 L) was added to the residue and the contents were stirred for about 1 h to obtain a solid product, which was filtered and washed with tert-butyl methyl ether (2 X 100 ml). The product was dried under vacuum below 60ºC to obtain the product as a 46.5 g dark brown compound, 46.5g ,yield 51%.
M.P.: 117-119oC; MS: 373(M+1); M.F.: C17H22F2N2O5..
Preparation of Intermediate -7: (5R)-Methanesulfonic acid 3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-2-oxo-oxazolidin-5-ylmethyl ester
To a mixture of (5R)-3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-5-hydroxymethyl-oxazolidin-2-one (45 g, 0.121 mol) in dichloromethane (0.3 L), was added triethylamine (24.5 g, 34 ml, 0.242 mol) while stirring. Methanesulfonyl chloride (18 g, 12.2 ml, 0.157 mol) was added to the above solution over a period of 1 h at 10oC -20oC and the reaction mixture was stirred for additional 2 h at RT. After completion of the reaction (TLC), the contents were evaporated under vacuum at 40oC to obtain an oily residue. Water (450 ml) was added to the residue and the traces of dichloromethane were removed under vacuum. The solid product thus obtained was filtered, washed with water (2 X 50 ml) and dried under vacuum at 70oC to obtain 50.6 g brownish compound. Yield = 93%; M.P.:106-108oC; MS: 451(M+1); M.F.: C18H24F2N2O7S.
Preparation of Intermediate 8a: (5R)-3-[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-5-azidomethyl-oxazolidin-2-one
Method A:
To a solution of (R)-3-(3,5-difluoro-4-(4-hydroxy-4-(methoxymethyl)piperidin-1-yl)phenyl)-5-(hydroxymethyl)oxazolidin-2-one (2g, 5.3 mmol),in tetrahydrofuran (20 mL), under argon , was added diphenylphosphoryl azide (1.63mL, 5.9 mmol). The solution was cooled to 0oC in an ice-bath. 1,8-diazabicyclo [5.4.0] undec-7-ene (DBU) (0.76mL, 4.9mmol) was added drop-wise over 15min..The reaction was stirred at same temperature for 1 hr, and then warmed to room temperature and stirred under for 16 hr. The reaction mixture was diluted with ethyl acetate (20 mL), and water (20mL). After separation of water layer, the organic layer was washed with water and 0.5M citric acid monohydrate (10 mL). The organic layer was dried over sodium sulfate and the solvent evaporated under reduced pressure.The residue was triturated with ether to obtain the product as a buff colored solid, 1.32g (62%).
M.P.: 106-108oC; M.S.- 398(M+1); M.F.- C17H21F2N5O4,
Method B:
To a solution of (5R)-methanesulfonic acid 3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-2-oxo-oxazolidin-5-ylmethyl ester (20 g, 0.044 mol, wet) in N,N-dimethylformamide (30 ml), was added sodium azide (8.6 g, 0.133 mol) in a single lot. The reaction mixture was gradually heated and the temperature was maintained at 70ºC for 8 h. After completion of the reaction (TLC), the contents were cooled to 20-25ºC and poured slowly into chilled water (300 ml). The solid product thus obtained was filtered and washed with water (2 x 50 ml). The wet product was air dried to obtain 16.5g dark brown compound (being an azide, it was NOT exposed to heat during drying) Yield ~ 93%.
M.P.: 106-108oC; MS : 398(M+1); M.F.: C17H21F2N5O4;:
Preparation of Intermediate 8b: (5S)-N-2-{3-[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-2-oxo-oxazolidin-5-ylmethyl}-phthalimide
Method A:
A mixture of (5R)-{3-[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)phenyl]-2-oxo-oxazolidin-5-yl methyl}-methanesulfonate(10g, 0.022 mol), Potassium phthalimide (12.2g, 0.066 mol) and DMF (50ml) was heated, with stirring, at 90oC for 4h. The resulting mixture was cooled to RT and poured over ice-water mixture. The separated solid was filtered, washed with water and dried under suction to obtain the product as a white solid, 9.46g, in 85% yield.
M.P.: 154-156 oC; MS: 502 (M+1); M.F. C25H25F2N3O6.
Method B:
To tetrahydrofuran (30 ml) were added triphenylphosphine (2.11g, 8 mmol)) and diethyldiazocarboxylate (1.62g, 8 mmol)), and the solution stirred at room temperature. After 10 minute phthalimide (1.18g, 8 mmol)) was added and after a further stirring for 10 minute, (R)-3-(3,5-difluoro-4-(4-hydroxy-4-(methoxymethyl)piperidin-1-yl)phenyl)-5-(hydroxymethyl) oxazolidin-2-one (2g, 5.3 mmol) was added and stirring continued further at room temperature. After 8 hrs ice-cold water (4 ml) was added to the reaction mixture and the resulting mixture was extracted by ethyl acetate (2 x 20ml). The ethyl acetate extract was dried (over sodium sulfate) and concentrated under reduced pressure. The residue was chromatographed on a column of silica gel to obtain the product as an off-white solid, 1.56g, yield 58%.
M.P.: 154-156 oC; MS : 502 (M+1); M.F. C25H25F2N3O6.
Preparation of Intermediate 10: (5S)- N-{3-[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide
via
Intermediate 9: 5-aminomethyl-3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-oxazolidin-2-one
Method A:
To a solution of (5R)-3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-5-azidomethyl-oxazolidin-2-one (10 g, 0.025 mol) in methanol (100 ml), were charged cobalt chloride (0.6 g, 0.0025 mol) followed by sodium borohydride (0.95 g, 0.025 mol) in small lots over a period of 30 minutes. The reaction mixture was stirred at RT for additional 2 h. After completion of the reaction , the contents were evaporated under vacuum below 40oC to obtain a sticky mass. The contents were suspended in a mixture of water (100 ml) and ethyl acetate (50 ml) and stirred for 15 minutes. The contents were filtered through a filter-aid bed and the bed was washed with ethyl acetate (2 X 25 ml). The layers were separated and the aqueous layer was further extracted with ethyl acetate (4 X 50 ml). The combined organic layer was washed with 1% HCl solution (100 ml). The aqueous layer was separated and washed with dichloromethane (4 X 50 ml). The pH of the aqueous layer was adjusted to 8 by adding saturated sodium bicarbonate solution. The contents were extracted with ethyl acetate (6 X 50 ml) till no amine spot was seen in the final organic extract. The combined organic layer (containing the intermediate 5-aminomethyl-3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-oxazolidin-2-one) was dried over anhydrous sodium sulfate.
Triethylamine (3.3 g, 4.5 ml, 0.0327 mol) was added to the above organic layer and acetyl chloride (2.17 g, 2 ml, 0.0277 mol) was added gradually over a period of 1 h at RT. The reaction mixture was stirred for 2 h and after completion of the reaction (TLC), the contents were washed with water (50 ml) and the layers separated. Activated carbon (1 g) was added to the organic layer and the contents were stirred for 15 minutes. The contents were filtered on a celite bed and the carbon-celite bed was washed with ethyl acetate (2 X 10 ml). The combined filtrate was evaporated under vacuum to obtain a slurry, which was filtered on a Buchner assembly and the product was washed with ethyl acetate (2 X 10 ml). The product was dried under vacuum at 70oC to obtain 5 g off-white solid. Yield = 48% (on the basis of azide). HPLC Purity ~ 98%.
M.P.: 178-179oC; MS : 414 (M+1); M.F.: C19H25F2N3O5.
Method B:
A solution of (5R)-3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-5-azidomethyl-oxazolidin-2-one (50 g, 0.125 mol) in ethyl acetatel (1L ml), were charged with 5g of 10% of Pd-C catalyst(50% wet) and the resulting mixture was hydrogenated at 30psi for 3h at 50oC.. The resulting mixture was cooled and filtered under suction over celite bed. The residue was washed with additional ethyl acetate (200ml). The combined filtrates were concentrated to 500ml volume.
To the above ethyl acetate solution was added Triethyl amine (19.1g, 0.189 mol), and acetic anhydride (16.1g, 1.58mol) in a single lot in few minutes). The reaction mixture was stirred for 16h at R.T. .The resulting mixture was cooled to 0-5oC, stirred for 0.5h and filtered under suction. The residue was washed with cold ethyl acetate(100ml) and dried at 70oC under reduced pressure to obtain the product as a a off-white solid, 43.5g, in 84% yield over two steps.
HPLC Purity ~ 98%
M.P.: 178-179oC; MS : 414 (M+1); M.F.: C19H25F2N3O5.
Method C:
To a solution of (S)-N-2-{3-[3,5-Difluoro-4-(4-methoxymethyl-4-hydroxypiperidine-1yl)phenyl]-2-oxo-oxazolidin-5-yl methyl}-phthalimide (2.77g, 0.0055mol) in ethanol (20ml) was added hydrazine hydrate ( 0.554g, 0.011mol) and the resulting solution stirred at RT for 6h. The solvent was evaporated under reduced pressure, the residue suspended in 3% sodium carbonate solution and extracted in dichloromethane (40ml). The dichloromethane layer was dried and to this solution was added triethylamine(1.11g, 0.011mol) and acetic anhydride (0.67g, 0.007mol) and the solution stirred for 6h at RT. The solvent was evaporated under reduced pressure and the residue purified by flash chromatography to obtain the product as white solid, 1.94g, in 85% yield.
M.P.: 178-179oC; MS: 414 (M+1); M.F.: C19H25F2N3O5.
Method D:
A mixture of (5R)-{3-[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)phenyl]-2-oxo-oxazolidin-5-yl methyl}-methanesulfonate (1gm, 4.4mmol) and sodium diformylamide (2gms, 22mmol) in DMF (5ml) was stirred at 95 ºC. for 15hrs. Then a mixture of conc. HCl (0.6ml) and water (0.6ml) and ethanol (8ml) were added. The solution was stirred at 75ºC for 5hrs. The mixture was concentrated under reduced pressure at 60-75 ºC. Water (1ml), ammonia solution (0.5ml) and acetic anhydride (1ml) was added to the residue and the mixture stirred at 70-75 ºC for 4-5 hrs. The solution was cooled to room temperature, diluted with water (5ml) and the separated solid filtered. The residue was washed with water (4ml.) and dried in a vacuum oven at 50ºC to obtain the product as an off-white solid, 0.37g, in 41% yield.
M.P.: 178-179oC; MS : 414 (M+1); M.F.: C19H25F2N3O5.
Method E:
To tetrahydrofuran (30 ml) were added triphenylphosphine (2.11g, 8 mmol)) and diethyldiazocarboxylate (1.62g, 8 mmol)), and the solution stirred at room temperature. After 10 min acetamide (0.475g, 8 mmol)) was added and after a further stirring for 10 min, (R)-3-(3,5-difluoro-4-(4-hydroxy-4-(methoxymethyl)piperidin-1-yl)phenyl)-5-(hydroxymethyl) oxazolidin-2-one (2g, 5.3 mmol) was added and stirring continued further at room temperature. After 16 hrs ice-cold water (4ml) was added to the reaction mixture and the resulting mixture was extracted by ethyl acetate (2 x 20ml). The ethyl acetate extract was dried (over sodium sulfate) and concentrated under reduced pressure. The residue was chromatographed on a column of silica gel to obtain the product as an off-white solid, 0.50g, yield 22%.
M.P.: 178-179oC; MS: 414 (M+1); M.F.: C19H25F2N3O5.
Preparation of Intermediate -11: (S)-N-{3-[3,5-Difluoro-4-(4-methoxymethyl-4-di-O-benzylphosphoryloxy-piperi din-1yl)-phenyl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide
To a solution of (S)-N-{3-[3,5-difluoro-4-(4-methoxymethyl-4-hydroxypiperidine-1yl)-phenyl]-2-oxo-oxazolidin-5-yl methyl}-acetamide (0.2 mmol) and tetrazole (0.6 mmol) in dichloromethane (5 ml) was added dibenzyl N,N,diisopropylphosphoramidite (0.4 mmol) and the resulting mixture was stirred for 4h. The resulting solution was cooled to 0 oC and 0.6 ml of 0.5M m-chloroperbenzoic acid solution in dichloromethane was added. After 4h, the solvent was evaporated under residue pressure and the residue chromatographed on a column of silica gel to obtain the product as a off-white solid in 75% yield,
MS: 674 (M+1); M.F. C33H38F2N3O8P;
Example A: Phosphoric acid mono-(1-{4-[(S)-5-(acetylamino-methyl)-2-oxo-oxazolidin-3-yl]-2,6-difluorophenyl}-4-methoxymethyl-piperidin-4-yl) ester
To a suspension of (S)-N-{3-[3,5-difluoro-4-(4-methoxymethyl-4-di-O-benzylphosphoryl- oxypiperidine-1yl)phenyl]-2-oxo-oxazolidin-5-yl methyl}-acetamide (0.15 mmol) and 20 % palladium hydroxide (20 mg) in 20 ml of a mixture of dichloromethane /aqueous methanol was stirred at room temperature for 6h. The catalyst was filtered and the residue evaporated under reduced pressure. The residue obtained was triturated with acetone to obtain a white solid as product in 70% yield.
MP. >140 °C; MS : 494(M+1) M.F.: C19H26F2N3O8P.
http://www.google.co.in/patents/WO2012059823A1?cl=en

 c) Converting intermediate of Formula (5) into intermediate of structure (6)
c) Converting intermediate of Formula (5) into intermediate of structure (6)


 f) Converting intermediate of Formula (11) into compound of Formula (A) or Pharmaceutically acceptable salts thereof
f) Converting intermediate of Formula (11) into compound of Formula (A) or Pharmaceutically acceptable salts thereof



 ormu a-
ormu a-
Scheme-1
Preparation of Intermediate 10: (5S)- N-{ 3-[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl- piperidin- 1 -yl)-phenyl] -2-oxo-oxazolidin-5-ylmethyl } -acetamide
via
Intermediate 9: 5-aminomethyl-3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-l- yl)-phenyl] -oxazolidin-2-one
Method A:
To a solution of (5R)-3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-l-yl)- phenyl]-5-azidomethyl-oxazolidin-2-one (10 g, 0.025 mol) in methanol (100 ml), were charged cobalt chloride (0.6 g, 0.0025 mol) followed by sodium borohydride (0.95 g, 0.025 mol) in small lots over a period of 30 minutes. The reaction mixture was stirred at RT for additional 2 h. After completion of the reaction , the contents were evaporated under vacuum below 40°C to obtain a sticky mass. The contents were suspended in a mixture of water (100 ml) and ethyl acetate (50 ml) and stirred for 15 minutes. The contents were filtered through a filter-aid bed and the bed was washed with ethyl acetate (2 X 25 ml). The layers were separated and the aqueous layer was further extracted with ethyl acetate (4 X 50 ml). The combined organic layer was washed with 1% HC1 solution (100 ml). The aqueous layer was separated and washed with dichloromethane (4 X 50 ml). The pH of the aqueous layer was adjusted to 8 by adding saturated sodium bicarbonate solution. The contents were extracted with ethyl acetate (6 X 50 ml) till no amine spot was seen in the final organic extract. The combined organic layer (containing the intermediate 5-aminomethyl-3-[3,5-difluoro-4-(4- hydroxy-4-methoxymethyl-piperidin-l-yl)-phenyl]-oxazolidin-2-one) was dried over anhydrous sodium sulfate.
Triethylamine (3.3 g, 4.5 ml, 0.0327 mol) was added to the above organic layer and acetyl chloride (2.17 g, 2 ml, 0.0277 mol) was added gradually over a period of 1 h at RT. The reaction mixture was stirred for 2 h and after completion of the reaction (TLC), the contents were washed with water (50 ml) and the layers separated. Activated carbon (1 g) was added to the organic layer and the contents were stirred for 15 minutes. The contents were filtered on a celite bed and the carbon-celite bed was washed with ethyl acetate (2 X 10 ml). The combined filtrate was evaporated under vacuum to obtain a slurry, which was filtered on a Buchner assembly and the product was washed with ethyl acetate (2 X 10 ml). The product was dried under vacuum at 70°C to obtain 5 g off-white solid. Yield = 48% (on the basis of azide). HPLC Purity ~ 98%.
M.P.: 178-179°C; MS : 414 (M+l); M.F.: C19H25F2N3O5. Method B:
A solution of (5R)-3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-l-yl)-phenyl]-5- azidomethyl-oxazolidin-2-one (50 g, 0.125 mol) in ethyl acetatel (1L ml), were charged with 5g of 10% of Pd-C catalyst(50% wet) and the resulting mixture was hydrogenated at 30psi for 3h at 50°C. The resulting mixture was cooled and filtered under suction over celite bed. The residue was washed with additional ethyl acetate (200ml). The combined filtrates were concentrated to 500ml volume. To the above ethyl acetate solution was added Triethyl amine (19. lg, 0.189 mol), and acetic anhydride (16. lg, 1.58mol) in a single lot in few minutes). The reaction mixture was stirred for 16h at R.T. .The resulting mixture was cooled to 0-5°C, stirred for 0.5h and filtered under suction. The residue was washed with cold ethyl acetate( 100ml) and dried at 70°C under reduced pressure to obtain the product as a a off-white solid, 43.5g, in 84% yield over two steps.
HPLC Purity ~ 98%
M.P.: 178-179°C; MS : 414 (M+l); M.F.: C19H25F2N3O5. Method C:
To a solution of (S)-N-2-{3-[3,5-Difluoro-4-(4-methoxymethyl-4-hydroxypiperidine- lyl)phenyl]-2-oxo-oxazolidin-5-yl methyl }-phthalimide (2.77g, 0.0055mol) in ethanol (20ml) was added hydrazine hydrate ( 0.554g, 0.01 lmol) and the resulting solution stirred at RT for 6h. The solvent was evaporated under reduced pressure, the residue suspended in 3% sodium carbonate solution and extracted in dichloromethane (40ml). The dichloromethane layer was dried and to this solution was added triethylamine(l.l lg, 0.01 lmol) and acetic anhydride (0.67g, 0.007mol) and the solution stirred for 6h at RT. The solvent was evaporated under reduced pressure and the residue purified by flash chromatography to obtain the product as white solid, 1.94g, in 85% yield.
M.P.: 178-179°C; MS: 414 (M+l); M.F.: C19H25F2N3O5. Method D:
A mixture of (5R)-{3-[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-l-yl)phenyl]- 2-oxo-oxazolidin-5-yl methyl }-methanesulfonate (lgm, 4.4mmol) and sodium diformylamide (2gms, 22mmol) in DMF (5ml) was stirred at 95 °C. for 15hrs. Then a mixture of cone. HC1 (0.6ml) and water (0.6ml) and ethanol (8ml) were added. The solution was stirred at 75°C for 5hrs. The mixture was concentrated under reduced pressure at 60-75 °C. Water (1ml), ammonia solution (0.5ml) and acetic anhydride (1ml) was added to the residue and the mixture stirred at 70-75 °C for 4-5 hrs. The solution was cooled to room temperature, diluted with water (5ml) and the separated solid filtered. The residue was washed with water (4ml.) and dried in a vacuum oven at 50°C to obtain the product as an off-white solid, 0.37g, in 41% yield.
M.P.: 178-179°C; MS : 414 (M+l); M.F.: C19H25F2N3O5. Method E:
To tetrahydrofuran (30 ml) were added triphenylphosphine (2.1 lg, 8 mmol)) and diethyldiazocarboxylate (1.62g, 8 mmol)), and the solution stirred at room temperature. After 10 min acetamide (0.475g, 8 mmol)) was added and after a further stirring for 10 min, (R)-3- (3,5-difluoro-4-(4-hydroxy-4-(methoxymethyl)piperidin-l-yl)phenyl)-5-(hydroxymethyl) oxazolidin-2-one (2g, 5.3 mmol) was added and stirring continued further at room temperature. After 16 hrs ice-cold water (4ml) was added to the reaction mixture and the resulting mixture was extracted by ethyl acetate (2 x 20ml). The ethyl acetate extract was dried (over sodium sulfate) and concentrated under reduced pressure. The residue was chromatographed on a column of silica gel to obtain the product as an off-white solid, 0.50g, yield 22%.
M.P.: 178-179°C; MS: 414 (M+l); M.F.: C19H25F2N3O5.
Wockhardt Research Center,
 IV
IV
 V
V
‘ Scheme-1 ‘
/////////
 THANKS AND REGARD'S
THANKS AND REGARD'S
DR ANTHONY MELVIN CRASTO Ph.D
GLENMARK SCIENTIST , NAVIMUMBAI, INDIA
did you feel happy, a head to toe paralysed man's soul in action for you round the clock
need help, email or call me
In this and the following Examples, “Formula III” refers to a compound wherein Z is
Figure US20100305069A1-20101202-C00024
and M=OH.
A 1-L, three-neck round-bottom flask equipped with a magnetic stirrer, nitrogen inlet/outlet and thermocouple was charged with the compound of Formula Ia below (16.0 g, 0.0499 mol], THF (320 mL, 20 vol) and Et3N (21.9 g, 0.216 mol, 5.0 equiv.).
Figure US20100305069A1-20101202-C00025
POCl3 (3.31 g, 0.0216 mol, 0.5 equiv.) was added dropwise via syringe over 5 minutes. The reaction temperature was maintained below 25° C. The batch was aged for 16 hours at room temperature at which point HPLC analysis (XBridge, C18) indicated that the reaction was complete. The reaction vessel was then immersed in an ice-water bath and a 500-mL addition funnel charged with 320 mL of H2O was attached to the reaction vessel. When the temperature of the reaction reached 2.7° C., H2O was added drop wise over 30 minutes. The temperature of the reaction was maintained below 10° C. Upon completion of the H2O addition, the ice-water bath was removed and the batch was aged for 3 hours. The solution was transferred to a 2-L round-bottom flask and concentrated under reduced pressure on a rotary evaporator. After removal of most of the THF from the solution, the aqueous mixture was extracted with 5 1-L portions of CH2Cl2:MeOH (9:1). The CH2Cl2 layers were combined and concentrated to a dark oil. This crude material was purified on 200 g of silica gel, eluting with 10% MeOH/CH2Cl2 to 20% 2 N NH3 in MeOH/CH2Cl2. Fractions containing mostly the bis-ester (as judged by TLC Rf=0.3 eluting with 20% 2 N NH3 in MeOH/CH2Cl2) were combined and concentrated under reduced pressure on a rotary evaporator, during which time a white precipitate was observed. The flask containing the slurry was removed from the rotary evaporator and equipped with a magnetic stir bar and allowed to stir while cooling to room temperature over 3 hours, during which time the slurry thickened. The solid was filtered and dried in a vacuum oven at 45° C. for 16 hours to give 3.55 g of bis-ester as an off-white solid (20% yield). HPLC analysis (Method A): 99.0% (AUC), tR=16.3 min. This reaction was repeated and the combined lots of the compound of Formula III (6.7 g) were slurried in 100 mL of MeOH (15 vol). The slurry was heated to 40° C. for 30 minutes and then allowed to cool to room temperature over 1 hour. The off-white solid was filtered and dried in a vacuum oven at 40° C. for 16 hours to give 6.15 g of the compound of Formula III (92% yield). The 1H NMR analysis of the product was consistent with the assigned structure. HPLC analysis (Method A): 99.0% (AUC), tR=16.3 min.
Example 2 Preparation of the Diphosphate Dihydrogen Diester, Formula IV
In Examples 2-5, “Formula IV” refers to a compound wherein Z is
Figure US20100305069A1-20101202-C00026
n=0 and M=O-imidazolium salt.
A 250-mL 3-neck round-bottom flask equipped with a magnetic stirrer, nitrogen inlet/outlet and thermocouple was charged with the compound of Formula IIa below (5.0 g, 11.1 mmol), carbonyldiimidazole (890 mg, 5.55 mmol, 0.5 equiv.) and DMF (100 mL, 20 vol).
Figure US20100305069A1-20101202-C00027
The suspension was heated to 50° C. and held at that temperature for 4 hours at which point HPLC analysis (XBridge, C18) indicated that the reaction was complete. The reaction was filtered at 50° C. and dried in a vacuum oven at 50° C. for 24 hours to give 5.15 g of the imidazolium salt (i.e., the compound of Formula IV) as an off-white solid (98% yield). The 1H NMR analysis of the product was consistent with the assigned structure. HPLC analysis (Method A): 94.5% (AUC), tR=14.6 min.
TABLE 1
Method A (Waters XBridge C18 Column)
Time (min) Flow (mL/min) % A % B
0.0 1.0 98.0 2.0
15.0 1.0 5.0 95.0
25.0 1.0 5.0 95.0
27.0 1.0 98.0 2.0
30.0 1.0 98.0 2.0
A = 87% 25 mM ammonium bicarbonate solution in water/13% Acetonitrile
B = Acetonitrile
Wavelength = 300 nm
disodium salt is TR 701..........................................
US8580767
Various oxazolidinone-containing compounds have been disclosed for use as antibiotics. For example, oxazolidinone-containing compounds have been described in U.S. patent application Ser. No. 10/596,412 (filed Dec. 17, 2004), and WO 04/048350, WO 03/022824 and WO 01/94342, which are incorporated herein by reference.
U.S. patent application Ser. No. 12/577,089 (filed Oct. 9, 2009) and U.S. patent application Ser. No. 12/699,864 (filed Feb. 3, 2010), which are assigned to the same assignee as in the present application, disclose phosphate dimer impurities made during the process of making of the compounds disclosed therein. Surprisingly, it has been found that compounds containing at least two phosphates binding two oxazolidinone-containing moieties, such as dimers of oxazolidinone-containing compounds have antibacterial activity similar to their dihydrogen monophosphate analog,
These active compounds have been disclosed in WO 05/058886 and US Patent Publication No. 20070155798, while processes for making these and related compounds have been disclosed in U.S. patent application Ser. No. 12/577,089 (filed Oct. 9, 2009), and a crystalline form of the phosphate ester and related salts of the above compound has been disclosed in U.S. patent application Ser. No. 12/699,864 (filed Feb. 3, 2010). The latter two applications are assigned to the same assignee as in the present application
.........................................................................................................................................
SYNTHESIS
US20070155798
DESCRIPTION OF COMPDS
10,
(R)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-5-hydroxymethyl oxazolidin-2-on (compound 10)
...........................................................................................................................................
18
Preparation of (R)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-5-fluoromethyl oxazolidin-2-on (compound 18)
...............................................................................................................................................................................
33
(R)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-5-methoxymethyl oxazolidin-2-on (compound 33)
..............................................................................................................................................................
59
(R)-[3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-2-oxo-5-oxazolidinyl]methyl disodiumphosphate (compound 59)
...........................................................................................................................................................................
72
mono-[(R)-[3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-2-oxo-5-oxazolidinyl]methyl]phosphate (compound 72)
COMPLETE SYNTHESIS
Example 5
Preparation of 2-cyano-5-bromopyridine
In 1 L of dimethylformamide was dissolved 100 g of 2,5-dibromopyridine, 32 g of cupper cyanide and 17.8 g of sodium cyanide were added to the solution at room temperature and the solution was stirred at the temperature of 150° C. for 7 hours for reaction. After being cooled to room temperature, the reaction mixture was added with water and extracted with ethyl acetate. The organic layer was washed with brine, dehydrated, filtered and concentrated in vacuo. The title compound 54 g was obtained. Yield 70%.
1HNMR(CDCl3) δ 8.76(s,1H), 7.98(dd,1H), 7.58(dd,1H)
Example 6
Preparation of 2-(tetrazol-5-yl)-5-bromopyridine
10 g of 2-cyano-5-bromopyridine prepared in the Preparation example 5 was dissolved in 100 ml of dimethylformamide, 5.33 g of sodiumazide, and 4.4 g of ammonium chloride were added to the solution at room temperature, and the solution was stirred at the temperature of 110° C. for 3 hours for reaction. The reaction mixture was added with water and then was extracted with ethyl acetate. The organic layer, thus separated, was washed with brine, dehydrated, filtrated and concentrated in vacuo thereby to obtain 10.5 g of the title compound. Yield 85%.
Preparation Example 7 Preparation of 2-(1-methyltetrazol-5-yl)-5-bromopyridine and 2-(2-methyltetrazol-5-yl)-5-bromopyridine
10.5 g of 2-(tetrazol-5-yl)-5-bromopyridine prepared in the Preparation example 6 was dissolved in 100 ml of dimethylformamide. And then 6.5 g of sodium hydroxide was added to the solution and 9.3 g of iodomethane was slowly added to the solution at the temperature of 0° C. The solution was stirred for 6 hours at room temperature, added with water, extracted with ethyl acetate. And then the organic layer was washed with brine, dehydrated, filtrated, concentrated in vacuo and purified by column chromatography to obtain 4 g of 2-(1-methyltetrazol-5-yl)-5-bromopyridine and 5 g of 2-(2-methyltetrazol-5-yl)-5-bromopyridine.
1) 2-(1-methyltetrazol-5-yl)-5-bromopyridine
1HNMR(CDCl3) δ 8.77(t,1H), 8.23(dd,1H), 8.04(dd,1H), 4.46(s,3H)
2) 2-(2-methyltetrazol-5-yl)-5-bromopyridine
1HNMR(CDCl3) δ 8.80(t,1H), 8.13(dd,1H), 7.98(dd,1H), 4.42(s,3H)
Example 1
Preparation of N-Carbobenzyloxy-3-fluoroaniline
3-fluoroaniline 100 g was dissolved in 1 L of tetrahydrofuran (THF) and the solution was added with 150 g (1.8 mol) of sodium bicarbonate (NaHCO3). After being cooled to 0° C., the solution was slowly added with 154 ml of N-carbobenzyloxy chloride (CbzCl) for reaction. While the temperature was maintained at 0° C., the reaction mixture was let to react for 2 hours with stirring. Afterwards, the reaction was extracted with 0.5 L of ethyl acetate. The organic layer, after being separated, was washed with brine, dried over anhydrous magnesium sulfate (MgSO4) and concentrated in vacuo. The residue was washed twice with n-hexane to afford the title compound as white crystal. 132 g. Yield 85%.
Example 2
Preparation of (R)-3-(3-fluorophenyl)-2-oxo-5-oxazolidinylmethanol
132 g of N-carbobenzyloxy-3-fluoroaniline 132 g prepared in the Preparation example 1 was dissolved in 1.3 L of tetrahydrofuran and the solution was cooled to −78° C. 370 ml of n-buthyllitium (n-BuLi, 1.6M/n-hexane) was slowly added to the solution in a nitrogen atmosphere, followed by stirring for 10 min. And 84 ml of (R)-(−)-glycidylbuthylate was slowly added to the reaction mixture, stirred at the same temperature for 2 hours and allowed to react for 24 hours at room temperature. After completion of the reaction, the solution was added with ammonium chloride (HH4Cl) solution and extracted with 0.5 L of ethyl acetate at room temperature. The organic layer, thus separated, was washed with brine, dried over anhydrous magnesium sulfate and concentrated in vacuo. The residue was dissolved in 100 ml of ethyl acetate and washed with n-hexane to give white crystals, which were purified to the title compound. 80 g. Yield 70%.
1H NMR (DMSO-d6) δ 7.85(t,1H), 7.58(dd,1H), 7.23(dd,1H), 4.69(m,1H), 4.02 (t,1H), 3.80(dd,1H), 3.60(br dd,2H).
Example 3
Preparation of (R)-3-(4-iodo-3-fluorophenyl)-2-oxo-5-oxazolidinylmethanol
In 300 ml of acetonitryl was dissolved 30 g of (R)-3-(3-fluorophenyl)-2-oxo-5-oxazolidinylmethanol prepared in the Preparation example 2, and 46 g of trifluoroacetic acid silver salt (CF3COOAg) and 43 g of iodide were added to the solution. After being stirred for one day at room temperature, the solution was added with water and was extracted with ethyl acetate. The organic layer, thus separated, was washed with brine and dehydrated. And then the residue was filtered, concentrated in vacuo and dried thereby to form the title compound 44 g. Yield 94%.
1H NMR (DMSO-d6) δ 7.77(t,1H), 7.56(dd,1H), 7.20(dd,1H), 5.20(m,1H), 4.70 (m,1H), 4.07(t,1H), 3.80(m,1H), 3.67(m,2H), 3.56(m,3H)
Example 4
Preparation of (R)-3-(4-tributhylstannyl-3-fluorophenyl)-2-oxo-5-oxazolidinylmethanol
In 660 ml of 1,4-dioxan was dissolved 50 g of (R)-3-(4-iodo-3-fluorophenyl)-2-oxo-5-oxazolidinylmethanol prepared in the Preparation example 3, 52 g of hexabutylditin ((Bu3Sn)2) and 9.3 g of dichlorobistriphenylphosphinpalladium were added into the solution, and stirred for 2 hours. The solution was filtered using celite and concentrated in vacuo. The residue was purified by column chromatography and 45 g of the title compound was formed.
1H NMR (DMSO-d6) δ 7.74(m,3H), 5.20(t,1H), 4.71(m,1H), 4.08(t,1H), 3.82(dd,1H), 3.68(m,1H), 3.52(m,1H), 1.48(m, 6H), 1.24(m, 6H), 1.06(m,6H), 0.83(t,9H)
COMPD 10
Example 1 Preparation of (R)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-5-hydroxymethyl oxazolidin-2-on (compound 10)
In 150 ml of 1-methyl-2-pyrrolidone was dissolved 37 g of (R)-3-(4-tributhylstannyl-3-fluorophenyl)-2-oxo-5-oxazolidinylmethanol. The solution was added with 19.7 g of 2-(2-methyltetrazol-5-yl)-5-bromopyridine, 10.44 g of lithium chloride and 2.9 g of dichlorobistriphenylphospine palladium(II) at room temperature and then stirred at the temperature of 120° C. for 4 hours. The reaction mixture was added with water and then extracted with ethyl acetate. The organic layer, thus separated, was washed with brine, dehydrated, filtrated, concentrated in vacuo and purified by column chromatography to provide 8 g of the title compound. Yield 26%.
1H NMR (DMSO-d6) δ 8.90(s,1H), 8.18(m,2H), 7.70(m,2H), 7.49(dd,1H), 5.25(t,1H), 4.74(m,1H), 4.46(s,3H), 4.14(t,1H), 3.88(dd,1H), 3.68(m,1H), 3.58 (m,1H)
COMPD 18
Example 28 Preparation of (R)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-5-fluoromethyl oxazolidin-2-on (compound 18)
In 5 ml of methylenchloride was dissolved 100 mg of the compound 10. The solution was added with 43 mg of diethylaminosulfurtrifloride (DAST) and 0.078 ml of triethylamine and then stirred for 24 hours. After being concentrating, the reaction mixture was purified by column chromatography to obtain the title compound 75 mg. Yield 75%.
1H NMR (DMSO-d6) δ 8.91(s,1H), 8.19(m,2H), 7.74(t,1H), 7.66(dd,1H) 7.49 (dd,1H), 5.06(m,1H), 4.89(m,2H), 4.46(s,3H), 4.23(t,1H), 3.95(dd,1H)
COMPD 33
Example 37 Preparation of (R)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-5-methoxymethyl oxazolidin-2-on (compound 33)
In 10 ml of methanol was dissolved 400 mg of (R)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-5-methansulfonyloxymethyl oxazolidin-2-on prepared in the secondary step of the Example 24. The solution was added with 90 mg of sodium methoxide at room temperature and then stirred for one day at room temperature. The solution was extracted with ethyl acetate and the organic layer, thus separated, was washed with water and brine. The organic layer was dehydrated, filtered, concentrated in vacuo and purified by column chromatography to provide the title compound 200 mg. Yield 58%.
1H NMR(CDCl3) δ 8.90(s,1H), 8.29(d,1H), 8.04(d,1H), 7.61(dd,1H), 7.58 (t,1H), 7.38(dd,1H), 4.80(m,1H), 4.45(s,3H), 4.08(t,1H), 3.96(dd,1H), 3.67 (m,2H), 3.43(s,3H)
COMPD 59
Example 58 Preparation of mono-[(R)-[3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-2-oxo-5-oxazolidinyl]methyl]phosphate (compound 72) and (R)-[3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-2-oxo-5-oxazolidinyl]methyl disodiumphosphate (compound 59)
1. The Primary Step
In 10 ml of mixture solvent (tetrahydrofuran:methylenchloride=1:1) was dissolved 1 g of compound 10. The solution was added with 0.6 g of tetrazole and 2.3 g of di-tetrabutyl diisoprophylphosphoamidite and stirred for 15 hours at room temperature. The reaction mixture was refrigerated to −78° C., added with 0.7 g of metachloroperbenzoic acid and stirred for 2 hours. After being cooling to −78° C., the reaction mixture was added with metachloroperbenzoic acid (0.7 g). When the reaction mixture was stirred for 2 hours, the temperature of the reaction mixture was raised to room temperature. The reaction mixture was then added with ethyl acetate. The organic layer, thus separated, was washed with sodium bisulfate, sodium bicarbonate and brine, dehydrated, filtered and concentrated in vacuo, followed by purification with column chromatography thereby to provide (R)-[3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-2-oxo-5-oxazolidinyl]methyl phosphoric acid ditetrabuthylester (0.71 g, 71%).
1H NMR (DMSO-d6) δ 8.90(s,1H), 8.18(m,2H), 7.74(t,1H), 7.68 (dd,1H), 7.49(dd,1H), 4.98(m,1H), 4.46(s,3H), 4.23(t,1H), 4.18(m,1H), 4.09(m,1H), 3.89 (dd,1H), 1.39(s,9H), 1.38(s,9H)
The crystal prepared the above method was dissolved in a mixture of methanol and chloroform. And then the solution added with 3.4 ml of sodium methoxide (0.3M methanol solution) at the room temperature and stirred for 10 hours. The reaction mixture was concentrated to prepare the residue. The residue was crystallized and filtered thereby to obtain the title compound (compound 59) 300 mg.
1H NMR (D2O) δ 8.27(s,1H), 7.56(dd,2H), 7.06(m,2H), 6.90(m,1H), 4.79 (m,1H), 4.63(s,3H), 3.90(m,4H)
COMPD 72
The Secondary Step
In 30 ml of methylenchloride was dissolved the compound (0.7 g) in the Primary Step. The solution was added with 15 ml of trifluoroacetic acid and then stirred for 1 hour at room temperature. The reaction mixture was concentrated in vacuo to prepare the residue. The residue was crystallized with ethanol and ethyl ether to obtain mono-[(R)-[3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-2-oxo-5-oxazolidinyl]methyl]phosphate (compound 72) 400 mg.
1H NMR (DMSO-d6) δ 8.92(s,1H), 8.20(m,2H), 7.74(t,1H), 7.66(dd,1H), 7.500(dd,1H), 4.95 (m,1H), 4.46(s,3H), 4.21(t,1H), 4.05(m,2H), 3.91(dd,1H)
US20070155798
...............................................................
IMPURITIES
US8426389
| Organic Impurities in TR-701 FA Drug Substance | |
| Impurity | |
| ‘Name’ | Structure and Chemical Name |
| Rx600013 ‘Des-methyl TR- 701’ |  |
| dihydrogen ((5R)-3-{3-fluoro-4-[6-(2H-1,2,3,4-tetrazol-5- | |
| yl)-3-pyridinyl]phenyl}-2-oxo-1,3-oxazolan-5-yl)methyl | |
| phosphate | |
| Rx600024 ‘Pyrophosphate’ |  |
| trihydrogen ((5R)-3-{3-fluoro-4-[6-(1-methyl-1H-1,2,3,4- | |
| tetraazol-5-yl)-3-pyridinyl]phenyl}-2-oxo-1,3-oxazolan-5- | |
| yl)methyl pyrophosphate | |
| Rx600014 ‘Ring opened’ |  |
| dihydrogen 3-{3-fluoro-4-[6-(2-methyl-2H-1,2,3,4-tetraazol-5- | |
| yl)-3-pyridinyl]aniline}-2-hydroxypropyl phosphate | |
| Rx600023 ‘Me-isomer’ |  |
| dihydrogen ((5R)-3-{3-fluoro-4-[6-(1-methyl-1H-1,2,3,4- | |
| tetraazol-5-yl)-3-pyridinyl]phenyl}-2-oxo-1,3-oxazolan-5- | |
| yl)methyl phosphate | |
| Rx600025 ‘Overalkylated- phosphorylated impurity’ |   |
| (R)-1-((3-(3-fluoro-4-(6-(2-methyl-2H-tetrazol-5- | |
| yl)pyridin-3-yl)phenyl)-2-oxooxazolidin-5-yl)methoxy)-3- | |
| hydroxypropan-2-yl dihydrogen phosphate; | |
| (R)-3-((3-(3-fluoro-4-(6-(2-methyl-2H-tetrazol-5- | |
| yl)pyridin-3-yl)phenyl)-2-oxooxazolidin-5-yl)methoxy)-2- | |
| hydroxypropyl dihydrogen phosphate | |
| Rx600020 ‘Dimer impurity’ |  |
| dihydrogen bis-O-O′-[(5R)-3-{3-fluoro-4-[6-(2-methyl- | |
| 2H-1,2,3,4-tetrazol-5-yl)-3-pyridinyl]phenyl}-2-oxo-1,3- | |
| oxazolidin-5-yl]methyl pyrophosphate | |
| Rx600026 “Chloro” |  |
| (R)-5-(chloromethyl)-3-(3-fluoro-4-(6-(2-methyl-2H- | |
| tetrazol-5-yl)pyridin-3-yl)phenyl)oxazolidin-2-one | |
| Rx600001 TR-700 |  |
| 5R)-3-{3-Fluoro-4-[6-(2-methyl-2H-1,2,3,4-tetrazol-5-yl)- | |
| pyridin-3-yl]-phenyl}-5-hydroxymethyl-1,3-oxazolidin-2-one | |
| Rx600022 ‘Bis phosphate’ |  |
| hydrogen bis-O-O′-[(5R)-3-{3-fluoro-4-[6-(2-methyl-2H-1,2,3,4- | |
| tetrazol-5-yl)-3-pyridinyl]phenyl}-2-oxo-1,3-oxazolidin-5-yl]methyl | |
| phosphate | |
| Rx600042 |  |
| 3-{[(5R)-3-{3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)pyridin-3- | |
| yl]phenyl}-2-oxo-1,3-oxazolidin-5-yl]methoxy}-2-hydroxypropyl | |
| [(5R)-3-{3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)pyridin-3- | |
| yl]phenyl}-2-oxo-1,3-oxazolidin-5-yl]methyl hydrogen phosphate | |
| Rx600043 |  |
| 2-{[(5R)-3-{3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)pyridin-3- | |
| yl]phenyl}-2-oxo-1,3-oxazolidin-5-yl]methoxy}-1-hydroxyethyl | |
| [(5R)-3-{3-fluoro-4-[6-(2-methyl-2H-tetrazol-5-yl)pyridin-3- | |
| yl]phenyl}-2-oxo-1,3-oxazolidin-5-yl]methyl hydrogen phosphate | |
| US4128654 | 10 Feb 1978 | 5 Dec 1978 | E. I. Du Pont De Nemours And Company | 5-Halomethyl-3-phenyl-2-oxazolidinones |
| US4250318 | 9 Aug 1978 | 10 Feb 1981 | Delalande S.A. | Novel 5-hydroxymethyl oxazolidinones, the method of preparing them and their application in therapeutics |
| US4340606 | 23 Oct 1980 | 20 Jul 1982 | E. I. Du Pont De Nemours And Company | 3-(p-Alkylsulfonylphenyl)oxazolidinone derivatives as antibacterial agents |
| US4461773 | 5 Jan 1984 | 24 Jul 1984 | E. I. Dupont De Nemours And Company | P-Oxooxazolidinylbenzene compounds as antibacterial agents |
| US4476136 | 24 Feb 1982 | 9 Oct 1984 | Delalande S.A. | Aminomethyl-5 oxazolidinic derivatives and therapeutic use thereof |
| US4948801 | 29 Jul 1988 | 14 Aug 1990 | E. I. Du Pont De Nemours And Company | Aminomethyloxooxazolidinyl arylbenzene derivatives useful as antibacterial agents |
| US5523403 | 22 May 1995 | 4 Jun 1996 | The Upjohn Company | Tropone-substituted phenyloxazolidinone antibacterial agents |
| US5565571 | 28 Apr 1994 | 15 Oct 1996 | The Upjohn Company | Substituted aryl- and heteroaryl-phenyloxazolidinones |
| US5652238 | 27 Sep 1994 | 29 Jul 1997 | Pharmacia & Upjohn Company | Esters of substituted-hydroxyacetyl piperazine phenyl oxazolidinones |
| US5688792 | 16 Aug 1994 | 18 Nov 1997 | Pharmacia & Upjohn Company | Substituted oxazine and thiazine oxazolidinone antimicrobials |
| US6365751 | 17 Apr 2001 | 2 Apr 2002 | Zeneca Ltd. | Antibiotic oxazolidinone derivatives |
| US6627646 * | 17 Jul 2001 | 30 Sep 2003 | Sepracor Inc. | Norastemizole polymorphs |
| US6689779 | 18 May 2001 | 10 Feb 2004 | Dong A Pharm. Co., Ltd. | Oxazolidinone derivatives and a process for the preparation thereof |
| US7129259 | 1 Dec 2004 | 31 Oct 2006 | Rib-X Pharmaceuticals, Inc. | Halogenated biaryl heterocyclic compounds and methods of making and using the same |
| US7141583 | 23 Apr 2001 | 28 Nov 2006 | Astrazeneca Ab | Oxazolidinone derivatives with antibiotic activity |
| US7144911 | 24 Dec 2003 | 5 Dec 2006 | Deciphera Pharmaceuticals Llc | Anti-inflammatory medicaments |
| US7202257 | 6 Jul 2004 | 10 Apr 2007 | Deciphera Pharmaceuticals, Llc | Anti-inflammatory medicaments |
| US7396847 | 9 Sep 2002 | 8 Jul 2008 | Astrazeneca Ab | Oxazolidinone and/or isoxazoline as antibacterial agents |
| US7462633 | 29 Jun 2004 | 9 Dec 2008 | Merck & Co., Inc. | Cyclopropyl group substituted oxazolidinone antibiotics and derivatives thereof |
| US7473699 | 25 Feb 2003 | 6 Jan 2009 | Astrazeneca Ab | 3-cyclyl-5-(nitrogen-containing 5-membered ring)methyl-oxazolidinone derivatives and their use as antibacterial agents |
| US7498350 | 24 Nov 2003 | 3 Mar 2009 | Astrazeneca Ab | Oxazolidinones as antibacterial agents |
| US7816379 | 17 Dec 2004 | 19 Oct 2010 | Dong-A Pharm. Co., Ltd. | Oxazolidinone derivatives |
| US20020115669 | 29 Aug 2001 | 22 Aug 2002 | Wiedeman Paul E. | Oxazolidinone chemotherapeutic agents |
| US20030166620 | 18 May 2001 | 4 Sep 2003 | Jae-Gul Lee | Novel oxazolidinone derivatives and a process for the preparation thereof |
| US20040180906 | 24 Dec 2003 | 16 Sep 2004 | Flynn Daniel L | Anti-inflammatory medicaments |
| US20050038092 | 29 Jun 2004 | 17 Feb 2005 | Yasumichi Fukuda | Cyclopropyl group substituted oxazolidinone antibiotics and derivatives thereof |
| US20050107435 | 9 Sep 2002 | 19 May 2005 | Gravestock Michael B. | Oxazolidinone and/or isoxazoline as antibacterial agents |
| US20050288286 | 6 Jul 2004 | 29 Dec 2005 | Flynn Daniel L | Anti-inflammatory medicaments |
| US20060116386 | 24 Nov 2003 | 1 Jun 2006 | Astrazeneca Ab | Oxazolidinones as antibacterial agents |
| US20060116400 | 24 Nov 2003 | 1 Jun 2006 | Astrazeneca Ab | Oxazolidinone and/or isoxazoline derivatives as antibacterial agents |
| US20060270637 | 24 Feb 2004 | 30 Nov 2006 | Astrazeneca Ab | Hydroxymethyl substituted dihydroisoxazole derivatives useful as antibiotic agents |
| US20070155798 | 17 Dec 2004 | 5 Jul 2007 | Dong-A Pharm. Co., Ltd. | Novel oxazolidinone derivatives |
| US20070185132 | 29 Jun 2004 | 9 Aug 2007 | Yasumichi Fukuda | Cyclopropyl group substituted oxazolidinone antibiotics and derivatives thereo |
| US20070191336 | 23 Dec 2004 | 16 Aug 2007 | Flynn Daniel L | Anti-inflammatory medicaments |
| US20070203187 | 22 Jan 2007 | 30 Aug 2007 | Merck & Co., Inc. | Cyclopropyl group substituted oxazolidinone antibiotics and derivatives thereof |
| US20070208062 | 24 May 2005 | 6 Sep 2007 | Astrazeneca Ab | 3-(4-(2-dihydroisoxazol-3-ylpyridin-5-yl)phenyl)-5-triazol-1-ylmethyloxazolidin-2-one derivatives as mao inhibitors for the treatment of bacterial infections |
| US20080021012 | 24 May 2005 | 24 Jan 2008 | Astrazeneca Ab | 3-[4-{6-Substituted Alkanoyl Pyridin-3-Yl}-3-Phenyl]-5-(1H-1,2,3-Triazol-1-Ylmethyl)-1,3-Oxazolidin-2-Ones As Antibacterial Agents |
| US20080021071 | 24 May 2005 | 24 Jan 2008 | Astrazeneca Ab | 3-{4-(Pyridin-3-Yl) Phenyl}-5-(1H-1,2,3-Triazol-1-Ylmethyl)-1,3-Oxazolidin-2-Ones as Antibacterial Agents |
| US20080064689 | 24 May 2004 | 13 Mar 2008 | Astrazeneca Ab | 3-[4-(6-Pyridin-3-Yl)-3-Phenyl] -5-(1H-1,2,3-Triazol-1-Ylmethyl)-1,3-Oxazolidin-2-Ones as Antibacterial Agents |
| US20090018123 | 19 Jun 2006 | 15 Jan 2009 | Milind D Sindkhedkar | Oxazolidinones Bearing Antimicrobial Activity Composition and Methods of Preparation |
| US20090192197 | 16 Sep 2008 | 30 Jul 2009 | Dong-A Pharm. Co., Ltd. | Novel oxazolidinone derivatives |
| US20100093669 | 9 Oct 2009 | 15 Apr 2010 | Trius Therapeutics | Methods for preparing oxazolidinones and compositions containing them |
| US20100227839 | 3 Feb 2010 | 9 Sep 2010 | Trius Therapeutics | Crystalline form of r)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin- 5-yl)-3-fluorophenyl)-5-hydroxymethyl oxazolidin-2-one dihydrogen phosphate |
| AU2004299413A1 | Title not available | |||
| AU2009200606A1 | Title not available | |||
| CA2549062A1 | 17 Dec 2004 | 30 Jun 2005 | Chong Hwan Cho | Novel oxazolidinone derivatives |
| CN101982468A | 17 Dec 2004 | 2 Mar 2011 | 东亚制药株式会社 | Novel oxazolidinone derivatives and pharmaceutical compositions comprising the derivatives |
| EP0312000A1 | 12 Oct 1988 | 19 Apr 1989 | The Du Pont Merck Pharmaceutical Company | Aminomethyl oxooxazolidinyl aroylbenzene derivatives useful as antibacterial agents |
| EP0352781A2 | 27 Jul 1989 | 31 Jan 1990 | The Du Pont Merck Pharmaceutical Company | Aminomethyloxooxazolidinyl arylbenzene derivatives useful as antibacterial agents |
| EP1699784A1 | 17 Dec 2004 | 13 Sep 2006 | Dong-A Pharmaceutical Co., Ltd. | Novel oxazolidinone derivatives |
| EP2305657A2 | 17 Dec 2004 | 6 Apr 2011 | Dong-A Pharmaceutical Co., Ltd. | Oxazolidinone derivatives |
| EP2435051A1 | 27 May 2010 | 4 Apr 2012 | Trius Therapeutics | Oxazolidinone containing dimer compounds, compositions and methods to make and use |
| IN236862A1 | Title not available | |||
| JPS5799576A | Title not available | |||
| KR20110071107A | Title not available | |||
| NZ547928A | Title not available | |||
| NZ575842A | Title not available | |||
| WO1993009103A1 | 5 Oct 1992 | 13 May 1993 | Upjohn Co | Substituted aryl- and heteroarylphenyloxazolidinones useful as antibacterial agents |
| WO1993023384A1 | 21 Apr 1993 | 25 Nov 1993 | Michael Robert Barbachyn | Oxazolidinones containing a substituted diazine moiety and their use as antimicrobials |
| WO1995007271A1 | 16 Aug 1994 | 16 Mar 1995 | Michael R Barbachyn | Substituted oxazine and thiazine oxazolidinone antimicrobials |
| WO1995014684A1 | 27 Sep 1994 | 1 Jun 1995 | Michel R Barbachyn | Esters of substituted-hydroxyacetyl piperazine phenyl oxazolidinones |
| WO2001094342A1 | 18 May 2001 | 13 Dec 2001 | Cho Jong Hwan | Novel oxazolidinone derivatives and a process for the preparation thereof |
| WO2002081470A1 | 3 Apr 2002 | 17 Oct 2002 | Astrazeneca Ab | Oxazolidinones containing a sulfonimid group as antibiotics |
| WO2003022824A1 | 9 Sep 2002 | 20 Mar 2003 | Astrazeneca Ab | Oxazolidinone and/or isoxazoline as antibacterial agents |
| WO2003035648A1 | 23 Oct 2002 | 1 May 2003 | Astrazeneca Ab | Aryl substituted oxazolidinones with antibacterial activity |
| WO2003047358A1 | 2 Dec 2002 | 12 Jun 2003 | Vaughan Leslie Crow | Cheese flavour ingredient and method of its production |
| WO2003072575A1 | 25 Feb 2003 | 4 Sep 2003 | Astrazeneca Ab | 3-cyclyl-5-(nitrogen-containing 5-membered ring) methyl-oxazolidinone derivatives and their use as antibacterial agents |
| WO2003072576A2 | 25 Feb 2003 | 4 Sep 2003 | Astrazeneca Ab | Oxazolidinone derivatives, processes for their preparation, and pharmaceutical compositions containing them |
| WO2004048350A2 | 24 Nov 2003 | 10 Jun 2004 | Astrazeneca Ab | Oxazolidinones as antibacterial agents |
| WO2004083205A1 | 16 Mar 2004 | 30 Sep 2004 | Astrazeneca Ab | Antibacterial 1, 3- oxazolidin -2- one derivatives |
| WO2005005398A2 | 29 Jun 2004 | 20 Jan 2005 | Yasumichi Fukuda | Cyclopropyl group substituted oxazolidinone antibiotics and derivatives thereof |
| WO2005051933A1 | 23 Nov 2004 | 9 Jun 2005 | Vijay Kumar Kaul | An improved process for the synthesis of 4-(4-benzyloxy-carbonylamino-2-fluorophenyl)-piperazine-1-carboxylic acid tert-butyl ester, a key intermediate for oxazolidinone antimicrobials and compounds prepared thereby |
| WO2005058886A1 | 17 Dec 2004 | 30 Jun 2005 | Dong A Pharm Co Ltd | Novel oxazolidinone derivatives |
| WO2005116017A1 | 24 May 2005 | 8 Dec 2005 | Astrazeneca Ab | Process for the preparation of aryl substituted oxazolidinones as intermediates for antibacterial agents |
| WO2006038100A1 | 6 Oct 2005 | 13 Apr 2006 | Ranbaxy Lab Ltd | Oxazolidinone derivatives as antimicrobials |
| WO2007023507A2 | 19 Jun 2006 | 1 Mar 2007 | Milind D Sindkhedkar | Oxazolidinones bearing antimicrobial activity composition and methods of preparation |
| WO2007138381A2 | 13 Oct 2006 | 6 Dec 2007 | Delorme Daniel | Phosphonated oxazolidinones and uses thereof for the prevention and treatment of bone and joint infections |
| WO2010042887A2 | 9 Oct 2009 | 15 Apr 2010 | Trius Therapeutics | Methods for preparing oxazolidinones and compositions containing them |
| WO2010091131A1 | 3 Feb 2010 | 12 Aug 2010 | Trius Therapeutics | Crystalline form of r)-3-(4-(2-(2-methyltetrazol-5-yl)pyridin-5-yl)-3-fluorophenyl)-5-hydroxymethyl oxazolidin-2-one dihydrogen phosphate |
| WO2010138649A1 | 27 May 2010 | 2 Dec 2010 | Trius Therapeutics, Inc. | Oxazolidinone containing dimer compounds, compositions and methods to make and use |
2 RADEZOLID
Radezolid
869884-78-6 cas no
869884-78-6, RX-103, RX-1741, RX-O1_667, Radezolid (USAN/INN), UNII-53PC6LO35W
Molecular Formula: C22H23FN6O3 Molecular Weight: 438.454823
Rib-X Pharmaceuticals
Phase II completed
N-{[(5S)-3-(2-fluoro-4′-{[(1H-1,2,3-triazol-5-ylmethyl)amino]methyl}biphenyl-4-yl)-2-oxo-1,3-oxazolidin-5-yl]methyl}acetamide
Rib-X Pharmaceuticals has completed two Phase II clinical trials of radezolid for the treatment of pneumonia and uncomplicated skin infections. The trial completion dates were in 2008 and 2009, but to date the Phase III trials have not been initiated [1-6].
Radezolid (INN, codenamed RX-1741) is a novel oxazolidinone antibiotic being developed by Rib-X Pharmaceuticals, Inc. for the treatment of serious multi-drug–resistant infections. Radezolid has completed two phase-II clinical trials. One of these clinical trials was for uncomplicated skin and skin-structure infections (uSSSI) and the other clinical trial was for community acquired pneumonia (CAP).
SCheme A

Scheme B illustrates the synthesis of intermediates 7 and 8 of the present invention using Suzuki coupling chemistry between boronic acids and aryl triflates. Boronic ester 6 is treated with an appropriate aryl triflate to yield the BOC-protected biaryl 7. The BOC group of 7 is removed to provide amine 8, an intermediate useful in the synthesis of certain compounds of the present invention.
Scheme B

8, R = NH2-HCI Scheme C depicts the synthesis of intermediates 9-13, which are useful in producing certain methoxy-substituted biaryl derivatives of the present invention. Suzuki coupling of boronic ester 6 produces biaryl aldehyde 9, which can be reduced to alcohol 10. Mesylation of 10 yields 11 that can be converted to azide 12. Reduction of azide 12 yields amine 13.
Scheme C

Scheme D depicts the synthesis of pyridyl intermediates, which are useful for the synthesis of compounds of the present invention, via similar chemistry to that shown in Scheme C. Coupling of boronic ester 6 to a halopyridine aldehyde produces biaryl aldehyde 14. Aldehyde 14 serves as the precursor to intermediates 15-18 via chemistry described above.
Scheme D

Biaryl aldehyde 19 (Scheme E) can be synthesized from a Suzuki coupling of iodide 1 and 4-formylphenylboronic acid. Scheme E illustrates how intermediate aldehydes of type 19, 9, and 14 can be converted via reductive amination chemistry to other amines, such as amines 20-22, which are useful as intermediates for the synthesis of certain compounds of the invention.
Scheme E

Scheme F depicts the general synthesis of compounds of type la and lb from amines of type 5, 13, 18, and 20-22. Compounds of type la and lb are synthesized via acylation of amines 5, 13 and 18 and 20-22 with the appropriate acids using, for example, l-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDCI) as the coupling agent. Compounds 4001-4007 were specifically synthesized from amine 5 and the appropriate carboxylic acids. Scheme F

Scheme G highlights the synthesis of compounds of general structure II from amines of type 5 and 18. The amine can be acylated with carboxylic acids using EDCI (or other commonly employed peptide coupling reagents known in the art) to afford amides II. Acid chlorides can be purchased or synthesized and allowed to react with amines 5 and 18, in the presence of bases such as triethylamine, to also produce amides II. Alternatively, carboxylic acids can be pre-loaded onto a solid polymeric support, such as a tetrafluorophenol containing resin (TFP resin), and reacted with amines to yield amide products of general structure II (such as compounds 4008-4015).
Scheme G

Scheme H illustrates the synthesis of compounds of general structure Ilia from amines of type 5, 13, and 18 using reductive amination chemistry. For example, biaryl amine compounds 4016-4028 are synthesized in this manner. Scheme H

Scheme I depicts the synthesis of general structure Illb of the present invention from amine intermediate 8. For example, compounds 4029-4031 are synthesized using this reductive amination chemistry.
Scheme I

Scheme J shows the synthesis of compounds of general structure IVa and IVb. Amines 20, 21, and 22 can be converted to tertiary amines IVa, such as compounds 4032-4034 and 4036, using standard reductive amination chemistry employed earlier for other derivatives. This reductive amination chemistry can be employed on biaryl aldehyde intermediates such as 19, 9, and 14 to yield optionally substituted amines of general structure IVb, illustrated by compound 4037.
Scheme J

producing compounds of the present invention. Known iodoaryl oxazolidinone intermediate 50 (see U.S. Patent Nos. 5,523,403 and 5,565,571) is coupled to a substituted aryl boronic acid (the Suzuki reaction) to produce biaryl alcohol 51. Mesylate 52, azide 53, and amine 54 are then synthesized using chemistry well known to those skilled in the art. Scheme 1

NaN3, DMF, 70 °C


http://www.google.co.il/patents/WO2005019211A2?hl=iw&cl=en
...................
| TABLE 1 | |
| Compound | |
| Number | Structure |
| 1 |  |
Example 1 Synthesis of Compound 1
Compound 1 and its hydrochloride salt are synthesized according to the following Scheme:


4-Methoxybenzyl Azide 1001. A solution of 4-methoxybenzyl chloride 1000 (51.8 g, 331.0 mmol) in anhydrous DMF (200 mL) was treated with solid sodium azide (21.5 g, 331.0 mmol, 1.0 equiv) at 25° C., and the resulting mixture was stirred at 25° C. for 24 h. When TLC and HPLC/MS showed that the reaction was complete, the reaction mixture was quenched with H2O (400 mL) and ethyl acetate (EtOAc, 400 mL) at room temperature. The two layers were separated, and the aqueous layer was extracted with EtOAc (200 mL). The combined organic extracts were washed with H2O (2×200 mL) and saturated NaCl aqueous solution (100 mL), dried over MgSO4, and concentrated in vacuo. The crude 4-methoxybenzyl azide (51.2 g, 53.95 g theoretical, 94.9% yield) was obtained as colorless oil, which by HPLC and 1H NMR was found to be essentially pure and was directly used in the subsequent reaction without further purifications. For 4-methoxybenzyl azide 1001: 1H NMR (300 MHz, CDCl3) δ 3.84 (s, 3H, ArOCH3), 4.29 (s, 2H, Ar—CH2), 6.96 (d, 2H, J=8.7 Hz), 7.28 (d, 2H, J=7.8 Hz).
C-[1-(4-Methoxy-benzyl)-1H-[1,2,3]triazol-4-yl]-methylamine and C-[3-(4-Methoxy-benzyl)-3H-[1,2,3]triazol-4-yl]-methylamine (1003 and 1004). A solution of 4-methoxybenzyl azide 1001 (61.2 g, 375.5 mmol) in toluene (188 mL) was heated with propargylamine 1002 (commercially available, 30.97 g, 38.6 mL, 563.0 mmol, 1.5 equiv) at 25° C., and the resulting reaction mixture was warmed up to gentle reflux at 100-110° C. for 21 h. When TLC and HPLC/MS showed that the reaction was complete, the reaction mixture was cooled down to room temperature before being concentrated in vacuo to remove the excess amount of propargylamine and solvent. The oily residue was then treated with 30% ethyl acetate-hexane (v/v, 260 mL), and the resulting mixture was warmed up to reflux and stirred at reflux for 30 min before being cooled down to room temperature for 1 h. The pale-yellow solids were then collected by filtration, washed with 30% ethyl acetate-hexane (v/v, 2×100 mL), and dried in vacuo at 40° C. for overnight to afford the crude, cycloaddition product (78.8 g, 81.75 g theoretical, 96.4%) as a mixture of two regioisomers, C-[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-yl]-methylamine and C-[3-(4-methoxy-benzyl)-3H-[1,2,3]triazol-4-yl]-methylamine (1003 and 1004), in a ratio of 1.2 to 1 by 1H NMR. The crude cycloaddition product was found to be essentially pure and the two regioisomers were not separated before being used directly in the subsequent reaction without further purification. For 1003 and 1004: 1H NMR (300 MHz, DMSO-d6) δ 1.82 (br. s, 2H, NH2), 3.72 and 3.73 (two s, 3H, Ar—OCH3), 5.47 and 5.53 (two s, 2H, ArCH2), 6.89 and 6.94 (two d, 2H, J=8.7 Hz, Ar—H), 7.17 and 7.29 (two d, 2H, J=8.7 Hz, Ar—H), 7.58 and 7.87 (two br. s, 1H, triazole-CH); C11H14N4O, LCMS (EI) m/e 219 (M++H) and 241 (M++Na).
4-({tert-Butoxycarbonyl-[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-phenylboronic acid and 4-({tert-Butoxycarbonyl-[3-(4-methoxy-benzyl)-3H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-phenylboronic acid (1008 and 1009). Method A. A solution of the regioisomeric C-[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-yl]-methylamine and C-[3-(4-methoxy-benzyl)-3H-[1,2,3]triazol-4-yl]-methylamine (1003 and 1004, 20.0 g, 91.74 mmol) in 1,2-dichloroethane (DCE, 280 mL) was treated with 4-formylphenylboronic acid 1005 (commercially available, 12.39 g, 82.57 mmol, 0.9 equiv) at room temperature, and the resulting reaction mixture was stirred at room temperature for 10 min. Sodium triacetoxyborohydride (NaB(OAc)3H, 29.2 g, 137.6 mmol, 1.5 equiv) was then added to the reaction mixture in three portions over the period of 1.5 h at room temperature, and the resulting reaction mixture was stirred at room temperature for an additional 3.5 h. When TLC and HPLC/MS showed that the reductive animation reaction was complete, the reaction mixture was concentrated in vacuo. The residue, which contained a regioisomeric mixture of 4-({[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-phenylboronic acid and 4-({[3-(4-methoxy-benzyl)-3H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-phenylboronic acid as the reductive animation products (1006 and 1007), was then treated with tetrahydrofuran (THF, 100 mL) and water (H2O, 100 mL). The resulting solution was subsequently treated with solid potassium carbonate (K2CO3, 37.98 g, 275.2 mmol, 3.0 equiv) and di-tert-butyl dicarbonate (BOC2O, 20.02 g, 91.74 mmol, 1.0 equiv) at room temperature and the reaction mixture was stirred at room temperature for 2 h. When TLC and HPLC/MS showed that the N-BOC protection reaction was complete, the reaction mixture was treated with ethyl acetate (EtOAc, 150 mL) and water (H2O, 100 mL). The two layers were separated, and the aqueous layer was extracted with ethyl acetate (50 mL). The combined organic extracts were washed with H2O (50 mL), 1.5 N aqueous HCl solution (2×100 mL), H2O (100 mL), and saturated aqueous NaCl solution (100 mL), dried over MgSO4, and concentrated in vacuo. The crude, regioisomeric 4-({tert-butoxycarbonyl-[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-phenylboronic acid and 4-({tert-butoxycarbonyl-[3-(4-methoxy-benzyl)-3H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-phenylboronic acid (1008 and 1009, 35.98 g, 37.32 g, 96.4%) was obtained as a pale-yellow oil, which solidified upon standing at room temperature in vacuo. This crude material was directly used in the subsequent reaction without further purification. For 1008 and 1009: 1H NMR (300 MHz, DMSO-d6) δ 1.32 and 1.37 (two br. s, 9H, COOC(CH3)3), 3.70, 3.73 and 3.74 (three s, 3H, Ar—OCH3), 4.07-4.39 (m, 4H), 5.49 and 5.52 (two s, 2H), 6.70-8.04 (m, 9H, Ar—H and triazole-CH); C23H29BN4O5, LCMS (EI) m/e 453 (M++H) and 475 (M++Na).
Method B. A solution of the regioisomeric C-[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-yl]-methylamine and C-[3-(4-methoxy-benzyl)-3H-[1,2,3]triazol-4-yl]-methylamine (1003 and 1004, 20.06 g, 92.0 mmol) in tetrahydrofuran (THF, 300 mL) was treated with 4-formylphenylboronic acid (13.11 g, 87.4 mmol, 0.95 equiv) at room temperature, and the resulting reaction mixture was stirred at room temperature for 10 min. Sodium triacetoxyborohydride (NaB(OAc)3H, 29.25 g, 138.0 mmol, 1.5 equiv) was then added to the reaction mixture in three portions over the period of 1.5 h at room temperature, and the resulting reaction mixture was stirred at room temperature for an additional 3.5 h. When TLC and HPLC/MS showed that the reductive animation reaction was complete, the reaction mixture, which contained a regioisomeric mixture of 4-({[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-phenylboronic acid and 4-({[3-(4-methoxy-benzyl)-3H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-phenylboronic acid as the reductive animation products (1006 and 1007), was then treated with water (H2O, 200 mL). The resulting aqueous solution was subsequently heated with solid potassium carbonate (K2CO3, 38.0 g, 276 mmol, 3.0 equiv) and di-tert-butyl dicarbonate (BOC2O, 20.08 g, 92 mmol, 1.0 equiv) at room temperature and the reaction mixture was stirred at room temperature for 2 h. When TLC and HPLC/MS showed that the N-BOC protection reaction was complete, the reaction mixture was treated with ethyl acetate (EtOAc, 150 mL) and water (H2O, 100 mL). The two layers were separated, and the aqueous layer was extracted with ethyl acetate (50 mL). The combined organic extracts were washed with H2O (50 mL), 1.5 N aqueous HCl solution (2×100 mL), H2O (100 mL), and saturated aqueous NaCl solution (100 mL), dried over MgSO4, and concentrated in vacuo. The crude, 4-({tert-butoxycarbonyl-[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-phenylboronic acid and 4-({tert-butoxycarbonyl-[3-(4-methoxy-benzyl)-3H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-phenylboronic acid (1008 and 1009, 38.45 g, 39.50 g, 97.3%) was obtained as a pale-yellow oil, which solidified upon standing at room temperature in vacuo. This crude material was found to be essentially identical in every comparable aspect as the material obtained from Method A and was directly used in the subsequent reaction without further purification.
(5S)-{4′-[5-(Acetylamino-methyl)-2-oxo-oxazolidin-3-yl]-2′-fluoro-biphenyl-4-ylmethyl}-[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-ylmethyl]-carbamic acid tert-butyl ester and (5S)-{4′-[5-(Acetylamino-methyl)-2-oxo-oxazolidin-3-yl]-2′-fluoro-biphenyl-4-ylmethyl}-[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-5-ylmethyl]-carbamic acid tert-butyl ester (1011 and 1012).
A suspension of the crude regioisomeric mixture of 4-({tert-butoxycarbonyl-[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-phenylboronic acid and 4-({tert-butoxycarbonyl-[3-(4-methoxy-benzyl)-3H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-phenylboronic acid (1008 and 1009, 37.62 g, 83.23 mmol) and N-[3-(3-fluoro-4-iodo-phenyl)-2-oxo-oxazolidin-5-ylmethyl]-acetamide (1010, 28.32 g, 74.9 mmol, 0.90 equiv) in toluene (150 mL) was treated with powder K2CO3 (34.45 g, 249.7 mol, 3.0 equiv), EtOH (50 mL), and H2O (50 mL) at 25° C., and the resulting mixture was degassed three times under a steady stream of Argon at 25° C. Pd(PPh3)4 (866 mg, 0.749 mmol, 0.01 equiv) was subsequently added to the reaction mixture, and the resulting reaction mixture was degassed three times again under a stead stream of Argon at 25° C. before being warmed up to gentle reflux for 18 h. When TLC and HPLC/MS showed the coupling reaction was complete, the reaction mixture was cooled down to room temperature before being treated with H2O (100 mL) and ethyl acetate (100 mL). The two layers were then separated, and the aqueous layer was extracted with EtOAc (100 mL). The combined organic extracts were washed with H2O (50 mL), 1.5 N aqueous HCl solution (2×150 mL), H2O (100 mL), and the saturated aqueous NaCl solution (100 mL), dried over MgSO4, and concentrated in vacuo. The residual oil was solidified upon standing at room temperature in vacuo to afford the crude, (5S)-{4′-[5-(acetylamino-methyl)-2-oxo-oxazolidin-3-yl]-2′-fluoro-biphenyl-4-y]methyl}-[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-ylmethyl]-carbamic acid tert-butyl ester (1011) and (5S)-{4′-[5-(acetylamino-methyl)-2-oxo-oxazolidin-3-yl]-2′-fluoro-biphenyl-4-ylmethyl}-[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-5-ylmethyl]-carbamic acid tert-butyl ester (1012) as a regioisomeric mixture. This crude product (43.36 g, 49.28 g theoretical, 88%) was used directly in the subsequent reaction without further purification. For the mixture of 1011 and 1012: 1H NMR (300 MHz, DMSO-d6) δ 1.35 and 1.38 (two br. s, 9H, COO(CH3)3), 1.85 (s, 3H, COCH3), 3.45 (t, 2H, J=5.4 Hz), 3.73 and 3.76 (two s, 3H, Ar—OCH3), 3.79 (dd, 1H, J=6.6, 9.1 Hz), 4.18 (t, 1H, J=9.1 Hz), 4.35-4.43 (m, 4H), 4.73-4.81 (m, 1H), 5.50 (br. s, 2H), 6.90 and 6.98 (two d, 2H, J=8.7 Hz), 7.28 and 7.32 (two d, 2H, J=8.7 Hz), 7.35 (dd, 2H, J=2.2, 8.6 Hz), 7.42 (dd, 1H, J=2.2, 8.6 Hz), 7.49-7.63 (m, 4H, aromatic-H), 7.90 and 7.99 (two br. s, 1H, triazole-CH), 8.29 (t, 1H, J=5.8 Hz, NHCOCH3); C35H39FN6O6, LCMS (EI) m/e 659 (M++H) and 681 (M++Na).
(5S)-N-{3-[2-Fluoro-4′-({[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-biphenyl-4-yl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide Hydrochloride (1013) and (5S)-N-{3-[2-Fluoro-4′-({[1-(4-methoxy-benzyl)-1H--[1,2,3]triazol-5-ylmethyl]-amino}-methyl)-biphenyl-4-yl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide Hydrochloride (1014). A solution of a regioisomeric mixture of (5S)-{4′-[5-(acetylamino-methyl)-2-oxo-oxazolidin-3-yl]-2′-fluoro-biphenyl-4-ylmethyl}-[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-ylmethyl]-carbamic acid tert-butyl ester and (5S)-{4′-[5-(acetylamino-methyl)-2-oxo-oxazolidin-3-yl]-2′-fluoro-biphenyl-4-ylmethyl}-[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-5-ylmethyl]-carbamic acid tert-butyl ester (1011 and 1012, 37.28 g, 56.65 mmol) in ethyl acetate (EtOAc, 150 mL) and methanol (MeOH, 30 mL) was treated with a solution of 4 N hydrogen chloride in 1,4-dioxane (113.3 mL, 453.2 mmol, 8.0 equiv) at room temperature, and the resulting reaction mixture was stirred at room temperature for 12 h. When TLC and HPLC/MS showed that the N-BOC deprotection reaction was complete, the solvents were removed in vacuo. The residue was then suspended in 250 mL of 5% methanol (MeOH) in acetonitrile (CH3CN), and the resulting slurry was stirred at room temperature for 1 h. The solids were then collected by filtration, washed with toluene (2×100 mL) and 5% methanol in acetonitrile (2×50 mL), and dried in vacuo to afford a regioisomeric mixture of the crude, (5S)-N-{3-[2-fluoro-4′-({[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-biphenyl-4-yl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide hydrochloride and (5S)-N-{3-[2-fluoro-4′-({[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-5-ylmethyl]-amino}-methyl)-biphenyl-4-yl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide hydrochloride (1013 and 1014, 30.0 g, 33.68 g theoretical, 89.1% yield) as off-white crystals in a ratio of 1.2 to 1. This material was found by 1H NMR and HPLC/MS to be essentially pure and was directly used in the subsequent reactions without further purification. For the regioisomeric mixture of 1013 and 1014: 1H NMR (300 MHz, DMSO-d6) δ 1.84 (s, 3H, COCH3), 3.44 (t, 2H, J=5.4 Hz), 3.71 and 3.74 (two s, 3H, Ar—OCH3), 3.80 (dd, 1H, J=6.6, 9.1 Hz), 4.17 (t, 1H, J=9.1 Hz), 4.23-4.30 (m, 4H), 4.73-4.80 (m, 1H), 5.58 and 5.70 (two s, 2H), 6.88 and 6.93 (two d, 2H, J=8.7 Hz), 7.15 and 7.32 (two d, 2H, J=8.7 Hz), 7.43 (dd, 2H, J=2.2, 8.6 Hz), 7.52-7.62 (m, 6H, aromatic-H), 8.28 (s, 1H, triazole-CH), 8.32 (t, 1H, J=5.8 Hz, NHCOCH3), 9.91 and 10.32 (two br. s, 2H, ArCH2N+H2); C30H31FN6O4, LCMS (EI) m/e 559 (M++H) and 581 (M++Na).
(5S)-N-[3-(2-Fluoro-4′-{[(1H-[1,2,3]triazol-4-ylmethyl)-amino]-methyl}-biphenyl-4-yl)-2-oxo-oxazolidin-5-ylmethyl]-acetamide hydrochloride (1 hydrochloride salt). A solution of the crude regioisomeric mixture of (5S)-N-{3-[2-fluoro-4′-({[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-4-ylmethyl]-amino}-methyl)-biphenyl-4-yl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide hydrochloride and (5S)-1H-{3-[2-fluoro-4′-({[1-(4-methoxy-benzyl)-1H-[1,2,3]triazol-5-ylmethyl]-amino}-methyl)-biphenyl-4-yl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide hydrochloride (1013 and 1014, 29.17 g, 49.07 mmol) in trifluoroacetic acid(TFA, 150 mL) was warmed up to 65-70° C., and the resulting reaction mixture was stirred at 65-70° C. for 12 h. When TLC and HPLC/MS showed that the deprotection reaction was complete, the solvents were removed in vacuo. The residual solids were then treated with ethyl acetate (EtOAc, 100 mL) and H2O (150 mL) before being treated with a saturated aqueous solution of sodium carbonate (30 mL) at room temperature. The resulting mixture was then stirred at room temperature for 1 h before the solids were collected by filtration, washed with EtOAc (2×50 mL) and H2O (2×50 mL), and dried in vacuo at 40-45° C. to afford the crude, (5S)-N-[3-(2-fluoro-4′-{[(1H-[1,2,3]triazol-4-ylmethyl)-amino]-methyl)-biphenyl-4-yl)-2-oxo-oxazolidin-5-ylmethyl]-acetamide (1 as the free base, 18.9 g, 21.49 g theoretical, 87.9%) as off-white powders, which by HPLC/MS and 1H NMR was found to be one pure regioisomer and this regioisomer was found to be identical as the material obtained from deprotection of 1013 alone by the same method. For 1 as the free base: 1H NMR (300 MHz, DMSO-d6) δ 1.85 (s, 3H, COCH3), 3.44 (t, 2H, J=5.4 Hz), 3.74 (s, 2H), 3.77 (s, 2H), 3.79 (dd, 1H, J=6.4, 9.2 Hz), 4.17 (t, 1H, J=9.1 Hz), 4.72-4.81 (m, 1H), 7.39-7.62 (m, 7H, aromatic-H), 7.73 (s, 1H, triazole-CH), 8.29 (t, 1H, J=5.8 Hz, NHCOCH3), 9.72 (br. s, 2H, ArCH2N+H2), 15.20 (br. s, 1H, triazole-NH); C22H23FN6O3, LCMS (EI) m/e 439 (M++H) and 461 (M++Na).
A suspension of 1 free base (18.0 g, 41.1 mmol) in ethyl acetate (EtOAc, 80 mL), and methanol (MeOH, 20 mL) was treated with a solution of 4.0 N hydrogen chloride in 1,4-dioxane (41.1 mL, 164.4 mmol, 4.0 equiv) at room temperature, and the resulting mixture was stirred at room temperature for 8 h. The solvents were then removed in vacuo, and the residue was further dried in vacuo before being treated with a mixture of 10% methanol in acetonitrile (80 mL). The solids were collected by filtration, washed with 10% MeOH/acetonitrile (2×40 mL), and dried in vacuo to afford 1 hydrochloride salt (18.13 g, 19.50 g theoretical, 93% yield) as off-white crystals.
The crude 1 hydrochloride salt can be recrystallized from acetonitrile and water, if necessary, according to the following procedure: A suspension of the crude 1 hydrochloride salt (50.0 g) in acetonitrile (1250 mL) was warmed up to reflux before the distilled water (H2O, 280 mL) was gradually introduced to the mixture. The resulting clear yellow to light brown solution was then stirred at reflux for 10 min before being cooled down to 45-55° C. The solution was then filtered through a Celite bed at 45-55° C., and the filtrates were gradually cooled down to room temperature before being further cooled down to 0-5° C. in an ice bath for 1 h. The solids were then collected by filtration, washed with acetonitrile (2×50 mL), and dried in vacuo at 40° C. for 24 h to afford the recrystallized 1 hydrochloride salt (42.5 g, 50.0 g theoretical, 85% recovery) as off-white crystals.
For 1: 1H NMR (300 MHz, DMSO-d6) δ 1.86 (s, 3H, COCH3), 3.45 (t, 2H, J=5.4 Hz), 3.84 (dd, 1H, J=6.4, 9.2 Hz), 4.19 (t, 1H, J=9.1 Hz), 4.24 (br. s, 2H), 4.31 (br. s, 2H), 4.74-4.79 (m, 1H), 7.44 (dd, 1H, J=2.2, 8.6 Hz), 7.57-7.66 (m, 6H, aromatic-H), 8.17 (s, 1H, triazole-CH), 8.30 (t, 1H, J=5.8 Hz, NHCOCH3), 9.72 (br. s, 2H, ArCH2N+H2), 15.20 (br. s, 1H, triazole-NH);
13C NMR (75 MHz, DMSO-d6) δ 22.57, 40.69, 41.50, 47.36, 49.23, 71.85, 105.70 (d, J=28.5 Hz), 114.14 (d, J=2.9 Hz), 122.29 (d, J=13.3 Hz), 128.82 (d, J=3.0 Hz), 130.70, 130.94, 131.0, 131.22, 135.30, 137.92 (br. s), 139.66 (d, J=11.2 Hz), 154.11, 159.13 (d, J=243.5 Hz), 170.19;
C22H23FN6O3—HCl, LCMS (EI) m/e 439 (M++H) and 461 (M++Na).
References
- Zhou J, Bhattacharjee A, Chen S, et al. (December 2008). "Design at the atomic level: design of biaryloxazolidinones as potent orally active antibiotics".Bioorg. Med. Chem. Lett. 18 (23): 6175–8. doi:10.1016/j.bmcl.2008.10.011. PMID 18947996.
- Skripkin E, McConnell TS, DeVito J, et al. (October 2008). "Rχ-01, a new family of oxazolidinones that overcome ribosome-based linezolid resistance".Antimicrob. Agents Chemother. 52 (10): 3550–7. doi:10.1128/AAC.01193-07. PMC 2565890. PMID 18663023.
- Lawrence L, Danese P, DeVito J, Franceschi F, Sutcliffe J (May 2008). "In vitro activities of the Rχ-01 oxazolidinones against hospital and community pathogens". Antimicrob. Agents Chemother. 52 (5): 1653–62. doi:10.1128/AAC.01383-07. PMC 2346622. PMID 18316525.
- Hanselmann R, Job G, Johnson G, Lou R, Martynow JG, Reeve MM (2009). "Synthesis of an antibacterial compound containing a 1,4-substituted 1H-1,2,3-triazole- a scaleable alternative to the "click" reaction"". Organic Process Research and Development 14: 152–158. doi:10.1021/op900252a.
- Franceschi F, Duffy EM (March 2006). "Structure-based drug design meets the ribosome". Biochem. Pharmacol. 71 (7): 1016–25.doi:10.1016/j.bcp.2005.12.026. PMID 16443192.
- Ohlsen K (November 2009). "Novel antibiotics for the treatment of Staphylococcus aureus". Expert Rev. Clin. Pharmacol. 2 (6): 661–72.doi:10.1586/ecp.09.26.
- Radezolid at Rib-X Pharmaceuticals
- http://www.unil.ch/webdav/site/cnfmi/shared/abstracts_and_lectures/2009/3__F_van_Bambeke.pdf
- Sutcliffe, J.A. Antibiotics in development targeting protein synthesis. Ann. NY Acad. Sci. 2011, 1241, 122–152, doi:10.1111/j.1749-6632.2011.06323.x.
- Rib-X. Radezolid. Available online: http://www.rib-x.com/pipeline/radezolid.php#development (accessed on 14 April 2013).
- Rib-X Pharmaceuticals, Inc. Safety and efficacy study of oxazolidinone to treat pneumonia. Available online: http://www.clinicaltrials.gov/ct2/show/NCT00640926 (accessed on 14 April 2013).
- Rib-X Pharmaceuticals, Inc. Safety and efficacy study of oxazolidinones to treat uncomplicated skin infections. Available online: http://www.clinicaltrials.gov/ct2/show/NCT00646958 (accessed on 14 April 2013).
- Shaw, K.J.; Barbachyn, M.R. The oxazolidinones: Past, present, and future. Ann. NY Acad. Sci. 2011, 1241, 48–70, doi:10.1111/j.1749-6632.2011.06330.x.
- Skripkin, E.; McConnell, T.S.; DeVito, J.; Lawrence, L.; Ippolito, J.A.; Duffy, E.M.; Sutcliffe, J.; Franceschi, F. Rχ-01, a new family of oxazolidinones that overcome ribosome-based linezolid resistance.Antimicrob. Agents Chemother. 2008, 52, 3550–3557, doi:10.1128/AAC.01193-07.
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US6969726 * | Jun 2, 2004 | Nov 29, 2005 | Rib X Pharmaceuticals Inc | Biaryl heterocyclic compounds and methods of making and using the same |
| US20050043317 * | Jun 2, 2004 | Feb 24, 2005 | Jiacheng Zhou | Biaryl heterocyclic compounds and methods of making and using the same |
9-17-2010
|
BIARYL HETEROCYCLIC COMPOUNDS AND METHODS OF MAKING AND USING THE SAME
| |
9-17-2010
|
Process for the synthesis of triazoles
| |
4-28-2010
|
BIARYL HETEROCYCLIC COMPOUNDS AND METHODS OF MAKING AND USING THE SAME
| |
11-26-2008
|
Biaryl heterocyclic compounds and methods of making and using the same
| |
10-26-2007
|
Method for reducing the risk of or preventing infection due to surgical or invasive medical procedures
| |
10-12-2007
|
Method for reducing the risk of or preventing infection due to surgical or invasive medical procedures
| |
10-12-2007
|
Method for reducing the risk of or preventing infection due to surgical or invasive medical procedures
| |
12-13-2006
|
Biaryl heterocyclic compounds and methods of making and using the same
| |
11-30-2005
|
Biaryl heterocyclic compounds and methods of making and using the same
|
October 10, 2012
QIDP Designation for Radezolid for Acute Bacterial Skin and Skin Structure Infections, Community-acquired Bacterial Pneumonia
Rib-X Pharmaceuticals announced that the FDA designated radezolid as a Qualified Infectious Disease Product (QIDP) for the indications of acute bacterial skin and skin structure infections (ABSSSI) and community-acquired bacterial pneumonia (CABP).
The QIDP designation will enable Rib-X to benefit from certain incentives for the development of new antibiotics, including an additional five years of market exclusivity, priority review and eligibility for fast-track status, provided under the new Generating Antibiotic Incentives Now (GAIN) program. GAIN was included in the FDA Safety and Innovation Act (FDASIA), formerly known as PDUFA V, which received bipartisan Congressional support and was signed into law by President Obama in July 2012.
Radezolid has completed two Phase 2 clinical trials with an oral formulation in uncomplicated skin and skin structure infections (uSSSI) and in CABP. A Phase 1 study with an IV formulation was recently completed in healthy subjects. Rib-X recently announced data from a positive Phase 1 IV dosing study conducted in healthy subjects and an in vivo long-term safety study vs. linezolid (Zyvox; Pfizer).
Radezolid is a next-generation oxazolidinone with a safety profile permitting long-term treatment of resistant infections, including those caused by methicillin-resistant Staphylococcus aureus (MRSA).
For more information call (203) 624-5606 or visit www.rib-x.com
3.............
CADAZOLID
CADAZOLID, ACT-179811
1-Cyclopropyl-6-fluoro-7-[4-({2-fluoro-4-[(5R)-5-(hydroxymethyl)-2-oxo-1,3-oxazolidin-3-yl]phenoxy}methyl)-4-hydroxypiperidin-1-yl]-4-oxo-1,4-dihydroquinolin-3-carboxylic acid
l-cyclopropyl-6-fluoro-7-{4-[2-fluoro-4-(R)-5-hydroxymethyl-2-oxo- oxazolidin-3-yl)-phenoxymethyl]-4-hydroxy-piperidin-l-yl}-4-oxo-l,4-dihydro- quinoline-3-carboxylic acid
| Formula | C29H29F2N3O8 |
|---|---|
| Mol. mass | 585.55 g/mol |
Actelion Pharmaceuticals Ltd / Actelion's novel antibiotic cadazolid receives US FDA Qualified Infectious Disease Product designation for the treatment of Clostridium difficile-associated diarrhea .
ALLSCHWIL/BASEL, SWITZERLAND - 27 February 2014 - Actelion Ltd (six:ATLN) today announced that the US Food and Drug Administration (FDA) has designated cadazolid as both a Qualified Infectious Disease Product (QIDP) and a Fast Track development program for the treatment of Clostridium difficile-associated diarrhea (CDAD).
The QIDP designation for cadazolid means that - among other incentives - cadazolid would receive a nine-month priority review upon successful completion of the ongoing global Phase III IMPACT program. The Fast Track designation is intended to promote communication and collaboration between the FDA and the Company on the development of the drug.
The designations are based on the 2012 US Generating Antibiotic Incentives Now (GAIN) Act. The GAIN act is a legislative effort to incentivize the development of new antibiotic agents that target serious life-threatening infections.
Guy Braunstein, M.D. and Head of Clinical Development commented: "Clostridium difficile-associated diarrhea is a very serious and potentially life-threatening infection. There is a great need for an antibiotic that allows effective treatment of CDAD with low recurrence rates, particularly in infections caused by hypervirulent strains. The GAIN act highlights the importance of research in this area and we are very happy to receive the advantages that this designation for cadazolid will afford us."
ABOUT THE IMPACT PROGRAM
IMPACT is an International Multi-center Program Assessing Cadazolid Treatment in patients suffering from Clostridium difficile-associated diarrhea (CDAD). The program comprises two Phase III studies comparing the efficacy and safety of cadazolid (250 mg administered orally twice daily for 10 days) versus vancomycin (125 mg administered orally four times daily for 10 days).
The IMPACT studies are designed to determine whether the clinical response after administration of cadazolid is non-inferior to vancomycin in subjects with CDAD, and whether administration of cadazolid is superior to vancomycin in the sustained clinical response. The program is expected to enroll approximately 1'280 subjects worldwide, and commenced enrollment in the fourth quarter of 2013.
ABOUT CADAZOLID
The novel antibiotic cadazolid is a strong inhibitor of Clostridium difficile protein synthesis leading to strong suppression of toxin and spore formation. In preclinical studies cadazolid showed potent in vitro activity against Clostridium difficile clinical isolates and a low propensity for resistance development. In a human gut model of CDAD, cadazolid had a very limited impact on the normal gut microflora.
Cadazolid absorption is negligible resulting in high gut lumen concentrations and low systemic exposure, even in severe cases of CDAD where the gut wall can be severely damaged and permeability to drugs potentially increased.
Cadazolid is an experimental antibiotic of the oxazolidinone class made by Actelion Pharmaceuticals Ltd. which is effective against Clostridium difficile, a major cause of drug resistant diarrhea in the elderly.[1] Current drug treatments for this infection involve orally delivered antibiotics, principally fidaxomicin, metronidazole and vancomycin; the last two drugs are the principal therapeutic agents in use, but fail in approximately 20 to 45% of the cases. The drug is presently in Phase III trials.[1] The drug works by inhibiting synthesis of proteins in the bacteria, thus inhibiting the production of toxins and the formation of spores.[2]
Structure
The chemical structure of cadazolid combines the pharmacophores of oxazolidinone and fluoroquinolone.[2]
In a study published in the journal Anaerobe, cadazolid has been shown to be effective in vitro against 133 strains of Clostridium difficile all collected from Sweden.[3]
In phase I tests, sixty four male patients reacted favourably to cadazolid which primarily acted and remained in the colon while displaying little toxicity even in regimes involving large doses.[1]
ABOUT CADAZOLID IN THE PHASE II STUDY
Cadazolid was studied in a Phase II multi-center, double-blind, randomized, active reference, parallel group, therapeutic exploratory study. The study evaluated the efficacy, safety and tolerability of a 10-day, twice daily oral administration of 3 doses (250 mg, 500 mg or 1,000 mg b.i.d.) of cadazolid in subjects with Clostridium difficile-associated diarrhea (CDAD). As the current standard of care for CDAD, oral vancomycin (125 mg qid for 10 days) was used as the active reference. The study was completed in December of 2012, after having enrolled 84 subjects with CDAD.
The results of the Phase II study indicate that the effect of all doses of cadazolid were numerically similar to, or better than vancomycin on key endpoints including CDAD clinical cure rates as well as sustained cure rates. Clinical cure rate was defined as the resolution of diarrhea and no further need for CDAD therapy at test-of-cure 24 to 72 hours after the last dose of treatment, while sustained cure rate was defined as clinical cure with no recurrence of CDAD up to 4 weeks post-treatment. Recurrence rates were numerically lower for all doses of cadazolid as compared to vancomycin. Cadazolid was safe and well tolerated.
ABOUT THE GAIN ACT (INCLUDING FAST TRACK DESIGNATION)
The Food and Drug Administration Safety and Innovation Act (FDASIA) was signed into law in July 2012. The GAIN Act is Title VIII to FDASIA. The purpose of the GAIN Act is to encourage pharmaceutical research of certain antibiotics by designation of products as QIDPs. These products are intended to treat serious or life-threatening infections and include those to treat certain specifically identified pathogens, which are listed in the GAIN Act. C. difficile is one such specifically identified pathogen and drugs to treat CDAD would be eligible for designation as a QIDP.
The GAIN Act also provides that qualifying drugs (QIDPs) are eligible for inclusion in the FDA's Fast Track program. This program is intended to facilitate development and expedite review of new drugs and includes close early communication between the FDA and a drug's sponsor.
ABOUT FAST TRACK DRUG DEVELOPMENT PROGRAMS
For further information regarding Fast Track Drug Development Programs, please refer to the FDA document "Guidance for Industry on Fast Track Drug Development Programs: Designation, Development, and Application Review". This document is available on the Internet at:
ABOUT CLOSTRIDIUM DIFFICILE-ASSOCIATED DIARRHEA
Clostridium difficile is a Gram-positive, anaerobic, spore-forming bacterium that is the leading cause of nosocomial diarrhea. Clostridium difficile-associated diarrhea (CDAD or CDI for Clostridium difficile infection) can be a severe and life-threatening disease and results from the overgrowth in the colon of toxigenic strains of Clostridium difficile, generally during or after therapy with broad-spectrum antibiotics. CDAD is a major healthcare problem and a leading cause of morbidity in elderly hospitalized patients. The frequency and severity of CDAD in the western world has increased in recent years, and new hypervirulent and epidemic strains of Clostridium difficile have been discovered that are characterized by overproduction of toxins and other virulence factors, and by acquired resistance to fluoroquinolones such as moxifloxacin.
Current antibiotic therapy for CDAD includes vancomycin and metronidazole. While clinical cure rates are generally 85-90%, recurrences rates of 15-30 % with either drug are problematic as Clostridium difficile produces spores that are resistant to antibiotic treatment and routine disinfection. Spores surviving in the gut of patients and/or in the hospital environment may play a major role in re-infection and recurrence of CDAD after antibiotic treatment. Vancomycin and metronidazole are reported to promote spore formation in vitro at sub-inhibitory concentrations.
Actelion Ltd.
Actelion Ltd. is a leading biopharmaceutical company focused on the discovery, development and commercialization of innovative drugs for diseases with significant unmet medical needs.
Actelion is a leader in the field of pulmonary arterial hypertension (PAH). Our portfolio of PAH treatments covers the spectrum of disease, from WHO Functional Class (FC) II through to FC IV, with oral, inhaled and intravenous medications. Although not available in all countries, Actelion has treatments approved by health authorities for a number of specialist diseases including Type 1 Gaucher disease, Niemann-Pick type C disease, Digital Ulcers in patients suffering from systemic sclerosis, and mycosis fungoides in patients with cutaneous T-cell lymphoma.
Founded in late 1997, with now over 2,400 dedicated professionals covering all key markets around the world including the US, Japan, China, Russia and Mexico, Actelion has its corporate headquarters in Allschwil / Basel, Switzerland
.......................
Preparation of the compound of formula II
The compound of formula II can be obtained by hydrogenation of the compound of formula VIII

VIII
over a noble metal catalyst such as palladium or platinum on charcoal in a solvent such as THF, MeOH or EA between 00C and 400C or by hydrolysis of in presence of a solution of HBr in water or AcOH between 00C and 800C in a solvent such as AcOH.
The compounds of formula III can be prepared as summarized in Scheme 1 hereafter.

IX VI IIIA: R1= H IIIS: ^ = SO2R5
Scheme 1
The compounds of formula V can be prepared as summarized in Scheme 2 hereafter.

II X XI

Scheme 2
The compounds of formula X can be prepared from the methylidene derivatives of formula XII as summarized in Scheme 3 hereafter.

Xc XII Xa: R1 = H

Scheme 3
Example 1:
l-cyclopropyl-6-fluoro-7-{4-[2-fluoro-4-((/f)-5-hydroxymethyl-2-oxo- oxazolidin-3-yl)-phenoxymethyl]-4-hydroxy-piperidin-l-yl}-4-oxo-l,4-dihydro- quinoline-3-carboxylic acid:
1 i. (R)-3-(3-fluoro-4-hydroxy-phenyl)-5-hydroxymethyl-oxazolidin-2-one:
A solution of (7?y)-3-(4-benzyloxy-3-fluoro-phenyl)-5-hydroxymethyl-oxazolidin-2-one (6.34 g, prepared according to WO 2004/096221) in THF/MeOH (1 :1; 200 ml) was hydrogenated over Pd/C 10% (1 g) overnight. The catalyst was filtered off, the filtrate evaporated under reduced pressure and the residue stirred in EA. The crystals were collected by filtration, affording 3.16 g (70% yield) of a colourless solid. 1H NMR (DMSOd6; δ ppm): 3.5 (m, IH), 3.64 (m, IH), 3.74 (dd, J = 8.8, 6.4, IH), 3.99 (t, J = 8.8, IH), 4.64 (m, IH), 5.16 (t, J = 5.6, IH), 6.93 (dd, J = 9.7, 8.8, IH), 7.08 (ddd, J = 8.8, 2.6, 1.2, IH), 7.45 (dd, J = 13.5, 2.6, IH), 9.66 (s, IH). MS (ESI): 228.1.
1. ii. 4-[2-fluoro-4- ((R)-5-hydroxymethyl-2-oxo-oxazolidin-3-yl)-phenoxymethyl]- 4-hydroxy-piperidine-l-carboxylic acid benzyl ester:
A solution of intermediate l.i (1.27 g) and l-oxa-6-aza-spiro[2.5]octane-6-carboxylic acid benzyl ester (1.60 g; prepared according to US 4244961) were dissolved in DMF (15 ml) and treated with Na2CO3 (1.16 g). The mixture was heated at 1000C overnight. The residue obtained after workup (DCM) was stirred in EA, and the solid was collected by filtration and sequentially washed with EA and Hex, affording 2.52 g (94.5% yield) of a beige solid.
1H NMR (DMSOd6; δ ppm): 1.57 (m, 4H), 3.14 (m, 2H), 3.54 (m, IH), 3.64 (m, IH), 3.79 (m, 5 H), 4.03 (t, J = 9.1, 1 H), 4.66 (m, 1 H), 4.78 (s, 1 H), 5.05 (s, 2 H), 5.16 (t,
J = 5.6, 1 H), 7.18 (m, 2 H), 7.32 (m, 5 H), 7.55 (d, J = 12, 1 H).
MS (ESI): 475.0.
1. iii. (R)-3-[3-fluoro-4-(4-hydroxy-piperidin-4-ylmethoxy)-phenyl]-5-hydroxymethyl- oxazolidin-2-one:
A suspension of intermediate l.ii (2.5 g) in EA/MeOH (1 :1; 100 ml) was hydrogenated over Pd/C for 48 h. The suspension was heated at 400C and the catalyst was filtered off.
The filtrate was evaporated under reduced pressure affording 1.61 g (89% yield) of a yellow powder.
1H NMR (DMSOd6; δ ppm): 1.4-1.63 (m, 4H), 2.67 (m, 2H), 2.83 (m, 2H), 3.53 (dd, J = 4.0, 12.0, IH); 3.66 (dd, J = 3.3, 12.0, IH), 3.71 (s, 2H); 3.80 (m, IH), 4.05 (t, J = 9.0,
IH), 4.48 (s, IH), 4.68 (m, IH), 5.20 (s, IH), 7.20 (m, 2H), 7.57 (d, IH).
MS (ESI): 341.5.
l.iv. l-cyclopropyl-6-fluoro-7-{4-[2-fluoro-4-((R)-5-hydroxymethyl-2-oxo-oxazolidin-3-yl)-phenoxymethyl]-4-hydroxy-piperidin-l-yl}-4-oxo-l,4-dihydro-quinoline-3-carboxylic acid:
A solution of intermediate l.iii (200 mg), 7-chloro-l-cyclopropyl-6-fiuoro-l,4-dihydro- 4-0X0-3 -quinolinecarboxylic acid boron diacetate complex (241 mg; prepared according to WO 88/07998) and DIPEA (100 μl) in NMP (2 ml) was stirred at 85°C for 5 h. The reaction mixture was evaporated under reduced pressure and the residue was taken up in 5M HCl in MeOH (3 ml) and stirred. The resulting solid was collected by filtration and washed with MeOH to afford 230 mg (67% yield) of a yellow solid.
1H NMR (DMSOd6; δ ppm): 1.66-1.35 (m, 4H), 1.75 (d, J = 12.8, 2H), 1.95 (m, 2H), 3.33 (t broad, J = 11.0, 2H), 3.57 (m, 3H), 3.67 (dd, J = 12.3, 3.3, IH), 3.83 (m, 2H), 3.92 (s, 2H), 4.06 (t, J = 9.0, IH), 4.69 (m, IH), 7.24 (m, 2H), 7.60 (m, 2H), 7.90 (d, J = 13.3, IH), 8.66 (s, IH).
MS (ESI): 585.9.
References
- Boschert, Sherry (19 Sep 2012). "Promising C. difficile Antibiotic in Pipeline". Internal Medicine News. International Medical News Group. Retrieved 22 May 2013.
- "Cadazolid". .actelion.com. Retrieved 2013-05-22.
- "Anaerobe - In vitro activity of cadazolid against Clostridium difficile strains isolated from primary and recurrent infections in Stockholm, Sweden". ScienceDirect.com. 2013-02-26. Retrieved 2013-05-22.
- WO 2008056335
- WO 2009136379
4.....................
Sutezolid
168828-58-8
N-({(5S)-3-[3-fluoro-4-(thiomorpholin-4-yl)phenyl]-2-oxo-oxazolidin-5-yl}methyl) acetamide
(S)—N-[[3-[3-fluoro-4-(4-thiomorpholinyl)phenyl]-2-oxo-5-oxazolidinyl]methyl]acetamide
Sutezolid, PNU-100480, U-100480, NSC742407, PNU 100480, 168828-58-8, Sutezolid [INN]
Molecular Formula: C16H20FN3O3S Molecular Weight: 353.41170
http://www.ama-assn.org/resources/doc/usan/sutezolid.pdf
Sutezolid (PNU-100480, PF-02341272) is an oxazolidinone antibiotic currently in development as a treatment for extensively drug-resistant tuberculosis.
Sutezolid, an antimicrobial oxazolidinone and the thiomorpholine analogue of linezolid, had been in early clinical development for the treatment of tuberculosis. However, development was discontinued.
The compound had been found to be active against Gram-positive bacteria such as multiresistant staphylococci, streptococci and enterococci. It was being developed by Pfizer. In 2011, orphan drug designation was assigned in the U.S. and the E.U. for the treatment of tuberculosis.
In 2013, Sequella acquired an exclusive worldwide license for the development and commercialization of sustezolid.
8-5-2011
|
Combination Therapy for Tuberculosis
|
Scheme 1 illustrates a general synthetic sequence for preparing compounds of the present invention.

Example 3 Preparation of (5S)-5-{[(4-chlorobenzylidene)amino]methyl}-3-(3-fluoro-4-thiomorpholin-4-ylphenyl)-1,3-oxazolidin-2-one
The title compound in Example 2 (194 g, 0.56 mole), and the title compound of Example 1 (195 g, 0.84 mole), and lithium tert-butoxide (116 g, 1.4 mole) were charged into a 3000 mL three neck round bottom flask under nitrogen. The reactants were slurried with methyl tert-butyl ether (1200 mL) and the mixture was warmed to 56° C. and stirred for 2 h as a yellow solid gradually formed. The reaction was cooled to room temperature, and diluted with 1200 mL water. The mixture was then stirred vigorously over 60 min as the solid changed from dark yellow to a more pale yellow solid. The mixture was cooled to 10° C., filtered, and the filter cake was washed with ice cold methyl tert-butyl ether (450 mL). The resulting light yellow solid was dried in air for 30 min, then placed in a vacuum oven and dried at 40° C. overnight to afford the title compound (243 g, 99% yield). 1H NMR (400 MHz, CDCl3): δ 2.8 (m, 4H), 3.2 (m, 4H), 3.9 (m, 2H), 4.1 (m, 2H), 5.0 (m, 1H), 6.9 (m, 1H), 7.2 (m, 1H), 7.4 (m, 3H), 7.6 (m, 2H), 8.4 (s, 1H).
Example 4 Preparation of N-{[(5S)-3-(3-fluoro-4-thiomorpholin-4-ylphenyl)-2-oxo-1,3-oxazolidin-5-yl]methyl}acetamide
The title compound in Example 3 (243 g, 0.56 mole) was combined with EtOAc (1300 mL) and water (1300 mL) in a 5000 mL three neck round bottom flask equipped with a mechanical stirrer. The mixture was treated drop-wise with 12N HCl (140 mL, 1.68 moles) and the mixture was stirred vigorously for 1 hour at room temperature. The layers were separated and the aqueous layer was washed with EtOAc (1×500 mL). The resulting aqueous solution containing (S)-5-(aminomethyl)-3-(3-fluoro-4-thiomorpholinophenyl)oxazolidin-2-one hydrochloride was combined with a mixture of dichloromethane (1800 mL) and MeOH (120 mL), and the vigorously stirred mixture was charged with acetic anhydride (132 mL, 1.4 mole) in one portion and subsequently treated drop-wise with 10 N NaOH (200 mL, 2.0 mole) over 15 min. An extremely thick reaction mixture resulted from addition of the base, which gradually thinned as the pH rose and the acylation rapidly progressed. The reaction was stirred vigorously for 1 hour after the mixture resolved to two phases. At that time, 10 M NaOH (160 mL, 1.6 mole) was added drop-wise to the mixture until the pH was stable at 7. The layers were separated, the aqueous layer was extracted with dichloromethane (250 mL), and the combined organic layers were dried over anhydrous potassium carbonate. The volatiles were removed in vacuo to give an off-white solid which was titrated with methyl tert-butyl ether (250 mL), collected, and dried in vacuo to give title compound (5) (186.1 g, 94% yield) as a fine white solid with greater than 98% HPLC purity (retention time=3.93 minutes, HPLC conditions reported below).
The crude solid was dissolved in warm 6% methanol in dichloromethane (1250 mL) in a 5000 mL three neck round bottom flask equipped with a mechanical stirrer. The solution was warmed to reflux, diluted by the portion-wise (500 mL) addition of 2500 mL isopropanol (IPA), and, in order to maintain reflux, the temperature was ramped to 50-70° C. On completion of this addition of IPA, the reflux condenser was replaced with a short-path distillation head and distillation was continued into a cooled flask. During distillation, a 500 mL portion of fresh IPA was added after 500 mL of distillate was collected to maintain between 2000 and 2500 mL IPA present at all times. After this addition (internal flask temperature dropped to 60° C.) the mixture became slightly cloudy and remained so for the balance of the distillation, becoming increasingly cloudy as the distillate temperature exceeded 70° C.; particulate matter appeared as the distillate temperature exceeded 75° C. The temperature controller was ramped to 85° C. and held there until the conclusion of the distillation. When the distillate was clearly isopropanol alone (82-83° C.) the volume was reduced to 2500 mL hot IPA, the heating mantle was removed, stirring was discontinued, and the paddle was removed from the flask. The mixture was allowed to continue to crystallize as the flask cooled. The white crystalline solid was then collected by filtration, washed with methyl tert-butyl ether (250 mL), and dried in vacuo at 40° C. to afford 180 g (91% yield) of the title compound in greater than 99% HPLC purity (retention time=3.93 minutes, HPLC conditions reported below). 1H NMR (400 MHz, DMSO-d6): δ 1.8 (s, 3H), 2.7 (m, 4H), 3.2 (m, 4H), 3.4 (m, 2H), 3.7 (m, 1H), 4.7 (m, 1H), 7.1 (m, 1H), 7.15 (m, 1H), 7.2 (m, 1H), 8.2 (m, 1H). Mass Spec. C16H20FN3O3S: m/z 354.1 (M+1).
HPLC conditions for analyses mentioned in the text: HP Series 1100; Column: Symmetry C8 5 uM 4.6×50 mm; Flow rate 1.2 mL/min; Solvent A: water with 0.1% formic acid, Solvent B: acetonitrile with 0.1% formic acid; Injection volume=10 uL of 1 mg/mL (acetonitrile); Gradient: Solvent B 0-100% over 7 minutes then 100% B for 1 minute; wavelength=254 nm.
Identification of a novel oxazolidinone (U-100480) with potent antimycobacterial activity
J Med Chem 1996, 39(3): 680
J Med Chem 1996, 39(3): 680
http://pubs.acs.org/doi/full/10.1021/jm950956y
(S)-N-[[3-[3-Fluoro-4-(4-thiomorpholinyl)phenyl]-2-oxo-5-oxazolidinyl]methyl]acetamide (6, U-100480). A solution of (R)-[3-[3-fluoro-4-(4-thiomorpholinyl)phenyl]-2-oxo-5-oxazolidinyl]methyl azide (19.662 g, 58.28 mmol) in dry THF (290 mL) was treated with triphenylphosphine (16.815 g, 64.11 mmol) over 10 min. After 2.0 h, TLC analysis (10% MeOH/CHCl3) revealed the conversion to iminophosphorane was complete. H2O (2.10 mL, 116.56 mmol) was added and the reaction mixture heated to 40 °C (internal temperature) for 5 h and then allowed to cool to ambient temperature overnight. At this point, TLC analysis (10% MeOH/CHCl3) indicated incomplete hydrolysis of the iminophosphorane intermediate. More H2O (8.40 mL) was added, and the reaction was heated to 40 °C for 5 h. At this time, TLC indicated complete conversion to the 5-(aminomethyl)oxazolidinone intermediate. The reaction mixture was first concentrated by rotary evaporation (benzene was added several times to azeotrope off the H2O) and then under high vacuum to give the crude amine as an off-white solid. This material was dissolved in CH2Cl2 (250 mL), treated with pyridine (46.099 g, 47.10 mL, 582.79 mmol) and acetic anhydride (29.749 g, 27.49 mL, 291.40 mmol), and then stirred overnight at ambient temperature. TLC analysis (10% MeOH/CHCl3) showed complete conversion to 6. The reaction mixture was diluted with CH2Cl2, transferred to a separatory funnel, and then washed with 1 N HCl until the washings were acidic. The organic layer was then washed with saturated aqueous NaHCO3 and brine, dried over Na2SO4, filtered, and concentrated in vacuo to give crude 6 (U-100480) as a cream-colored solid. The crude product was triturated with hot CHCl3; most but not all of the solids dissolved. After cooling to ambient temperature, the solids were filtered off (cold CHCl3 wash) and dried in vacuo to furnish 13.174 g of analytically pure title compound as a white solid. A second crop of 3.478 g, also analytically pure, afforded a combined yield of 81%:
mp 186.5−187.0 oC; [α]D −8° (c 1.00, CHCl3);
IR (mull) 1749, 1746, 1641, 1656, 1518, 1448, 1419, 1225, 1215, 1158, 1106, 1083, 867 cm-1;
1H NMR (300 MHz, CDCl3) δ 7.42 (dd, 1H, J = 2.6, 14.0 Hz), 7.06 (ddd, 1H, J = 1.0, 2.6, 8.8 Hz), 6.95 (dd, 1H, J = 9.0, 9.0 Hz), 6.61 (br t, 1H, J = 6.0 Hz), 4.81−4.72 (m, 1H), 4.02 (dd, 1H, J = 9.0, 9.0 Hz), 3.75 (dd, 1H, J = 6.7, 9.1 Hz), 3.71−3.55 (m, 2H), 3.32−3.27 (m, 4H), 2.84−2.79 (m, 4H), 2.02 (s, 3H);
MS m/z (rel intensity) 353 (M+, 100), 309 (31), 279 (5), 250 (17), 235 (14), 225 (20), 212 (7), 176 (19), 138 (18), 42 (28);
HRMS calcd for C16H20N3O3FS 353.1209, found 353.1200. Anal. (C16H20N3O3FS) C, H, N.
see aLSO
WO 1995007271
WO 2010026526
Repurposed drugs for tuberculosis treatment.
5.........................
EPEREZOLID
EPEREZOLID
pfizer.originator
Eperezolid [USAN], PNU 100592, U-100592,
Molecular Formula: C18H23FN4O5
Molecular Weight: 394.397423
(S)-N-[[3-[3-Fluoro-4-[4-(2-hydroxyacetyl)piperazin-1-yl]phenyl]-2-oxo-1,3-oxazolidin-5-yl]methyl]acetamide
(S)-N-[[3-[3-fluoro-4-[4-(hydroxyacetyl)-l-piperazinyl]- phenyl]-2-oxo-5-oxazolidinyl]methyl]acetamide
Oxazolidinones are a new class of Gram-positive antibacterial agents which are known to those skilled in the art, see for example US 5,688,792. (S)-N-[[3-[3- fluoro-4-(4-morpholinyl)phenyl]-2-oxo-5-oxazolidinyl]methyl]acetamide, known as linezolid, the compound of Example 5 of US Patent 5,688,792 is known and has the following chemical formula:

(S)-N-[[3-[3-fluoro-4-[4-(hydroxyacetyl)-l-piperazinyl]-phenyl]-2-oxo-5- oxazolidinyl]methyl]acetamide, known as eperezolid, the compound of
Example 8 of US Patent 5,837,870 is known and has the following chemical formula:

Linezolid and eperezolid can be produced by the processes set forth in US Patents 5,688,791 and 5,837,870 as well as that of International Publication WO99/24393. It is preferably produced by the process of US Patent 5,837,870.
It is preferred that the linezolid produced be used in crystal form π, which has the characteristics set forth in CHART A. Once linezolid is synthesized, crystal Form π is prepared by starting with linezolid of high enantiomeric purity. It is preferred that the linezolid be more than 98% enantiomerically pure, it is more preferred that the linezolid be more than 99% pure and it is even more preferred that the linezolid be 99.5% pure. The linezolid of greater than 98% enantiomeric purity to be used to form crystal form II can either be in solution or be a solid. The linezolid starting material, solid or solution, is mixed with a solvent selected from the group consisting of compounds of the formula: water, acetonitrile, chloroform, methylene chloride, R OH where R\ is Cι-C6 alkyl; Rι-CO-R2 where R2 is Cι-C alkyl and Ri is as defined above; phenyl substituted with 1 thru 3 Ri where Ri is as defined above; Rι-CO-O-R2 where Ri is -C alkyl and Ri is as defined above; Rι-O-R2 where
is Cι-C6 alkyl and Ri is as defined above. It is preferred that the solvent be selected from the group consisting of water, ethyl acetate, methanol, ethanol, propanol, isopropanol, butanol, acetonitrile, acetone, methyl ethyl ketone, chloroform, methylene chloride, toluene, xylene, diethyl ether, or methyl-t-butyl ether. It is more preferred that the solvent be ethyl acetate, acetone, acetonitrile, propanol, or isopropanol. It is most preferred that the solvent be ethyl acetate. The mixture of linezolid in the solvent is agitated at a temperature below 80° until crystals of Form II are formed and crystals of other solid forms, such as Form I, disappear. It is preferred to dissolve the linezolid in ethyl acetate at a temperature near the boiling point of the solvent. This mixture is cooled to a temperature of about 70°. The mixture may be seeded with crystals of Form II to facilitate crystallization. It is preferred that the solid product is cooled and agitated at a temperature between about 45° and about 60° until the solids consist only of Form II crystals. It is most preferred to maintain the slurry at a temperature of about 55°. It is preferred to mix the linezolid and solvent for at least 10 min, it is even more preferred to mix the linezolid and solvent for at least 20 min and it is most preferred to mix the linezolid and solvent for at least 30 min. The time and temperature will vary depending on the solvent selected. With ethyl acetate it is preferred to mix for not less that 60 minutes. The crystalline slurry may be further cooled to improve yield, and the solid Form II product may be isolated. The mixture may be further cooled and agitated. Other measures which can be used to facilitate crystallization include, but are not limited to, cooling, concentration of the solution by evaporation or distillation, or through addition of other solvents. The crystals are isolated by procedures known to those skilled in the art.
It is well known to those skilled in the art that the oxazolidinones are useful as anti-bacterial agents especially against Gram-positive organisms. US Patent 5,688,792 discloses that oxazolidinones can be administered IV. The preferred formulation for linezolid IV solution is: Linezolid 2.0 mg mL
Sodium Citrate Dihydrate (USP) 1.64 mg/mL
Citric Acid Anhydrous (USP) 0.85 mg/mL
Dextrose Monohydrate (USP) 50.24 mg/mL
Hydrochloric Acid ( 10%) q.s. to pH 4.8 (pH 4.6 to 5.0) Sodium hydroxide (10%) q.s. to pH 4.8 (pH 4.6 to 5.0)
Water for Injection (USP) q.s. ad 1.0 mL
The linezolid IV solution is formulated by heating water for injection from about 50 to about 65°. Next the sodium citrate, citric acid and dextrose are added and stirred until dissolved. An aqueous slurry of linezolid is added to the previous mixture and stirred until dissolved. The mixture is cooled to 25° with stirring. The pH is measured and adjusted if necessary. Last the mixture is brought to volume, if necessary, with water for injection. The mixture is filtered, filled into infusion containers, over wrapped and terminally moist heat sterilized.
The aqueous solution for IV administration can be placed in the container which is selected from the group consisting of a bag, a bottle, a vial, a large volume parenteral, a small volume parenteral, a prefilled syringe and a cassette. It is realized that a vial is a bottle. However, those skilled in the art use the term "bottle" to refers to larger bottles and "vials" to refer to smaller bottles. It is preferred that the container be a bag, a bottle, a vial or a prefilled syringe. It is more preferred that the container be a bag or bottle. It is most preferred that the container be a bag. The shape and/or size of the container is unimportant. It is preferred that the container be a bag sufficient to hold 25 to 2,000 mL of IV solution. It is preferred that the linezolid mixture be put in bags in amounts of 100, 200 or 300 mL of solution however smaller or larger volumes are acceptable.
.................
. Scheme 2. Synthesis of eperezolid

RR==IH-

s

Et3N,



17 (eperezolid)
1-(2-Fluoro-4-nitrophenyl)piperazine (8). To 3,4-difluoronitrobenzene (20.5 g, 129 mmol) in acetonitrile (290 mL) was added triethylamine (36 mL) and piperazine (32 g, 387 mmol). The mixture was stirred at reflux for 18 h, after which it was cooled to room temperature and partitioned between H2O (500 mL) and EtOAc (400 mL). The layers were separated and the aqueous layer was extracted with EtOAc (2 x 300 mL). The organic layers were combined and washed with saturated NaCI solution (400 mL). The saturated NaCI layer was extracted again with EtOAc (2 x 200 mL). The organic layers were combined, dried over Na2SO4, filtered and concentrated to yield 8 as a yellow solid (29 g, quant.). 1H NMR (400 MHz, CDCI3) δ 1.63 (s, 1 H), 3.04-3.06 (m, 4H), 3.25-3.28 (m, 4H), 6.91 (t, J=8.7, 1 H), 7.90 (dd, J=13.2, 2.5, 1 H), 7.97- 8.00 (m, 1H).
3-Fluoro-4-(piperazin-1-yl)benzenamine (9). Compound 8 (10.0 g, 44.4 mmol) was dissolved in anhydrous EtOH (222 mL) and placed in a Parr pressure flask. PtO2 catalyst (31 mg) was added and the mixture was agitated under 50-60 psi of H2 on a Parr apparatus for 30 min, after which the reaction mixture was vented, more catalyst was added (78 mg) and the reaction mixture was submitted to 50-60 psi of H2 for another 30 min. The reaction mixture was filtered on Celite, the solid was washed with MeOH1 and the combined filtrates were concentrated to give 9 as a yellow solid (8.7 g, quant.). 1H NMR (400 MHz, CDCI3) δ 1.64 (bs,
1 H), 2.92-2.94 (m, 4H), 3.02-3.04 (m, 4H), 5.53 (bs, 2H)1 6.38-6.45 (m, 2H), 6.80 (t, J=8.5, 1 H).
Benzyl 4-(4-((benzyloxy)carbonyl)piperazin-1 -yl)-3-fluorophenylcarbamate (10).
Compound 10 was obtained in 78% yield (light yellow solid) using the protocol described in J. Med. Chem. 1996, 39, 673-679. 1H NMR (400 MHz, CDCI3) δ 2.98 (bs, 4H), 3.65-3.68 (m, 4H),
5.16 (s, 2H), 5.19 (s, 2H), 6.59 (bs, 1H), 6.85 (t, J=9.1 , 1 H), 6.94-6.97 (m, 1 H), 7.27-7.41 (m,
11H).
Benzyl 4-(2-fluoro-4-((R)-5-(hydroxymethyl)-2-oxo-oxazolidin-3-yl)phenyl) piperazine-1-carboxylate (11). Compound 11 was obtained in 66% yield (off-white solid) using the protocol described in J. Med. Chem. 1996, 39, 673-679. 1H NMR (400 MHz, CDCI3) δ 3.01
(bs, 4H), 3.66-3.69 (m, 4H), 3.74-3.79 (m, 1H)1 3.92-4.03 (m, 3H), 4.71-4.77 (m, 1H), 5.16 (s,
2H), 6.91 (t, J=9.1 , 1 H), 7.11-7.14 (m, 1H), 7.91-7.38 (m, 5H), 7.46 (dd, J=14.2, 2.5, 1 H).
Benzyl 4-(2-fluoro-4-((/?)-5-(methanesulfonyloxymethyl)-2-oxo-oxazolidin-3- yl)phenyl) piperazine-1-carboxylate (12). Compound 12 was obtained in quantitative yield (off- white foam) using the protocol described in J. Med. Chem. 1996, 39, 673-679. 1H NMR (400 MHz, CDCI3) δ 3.02 (bs, 4H), 3.10 (s, 3H), 3.67-3.69 (m, 4H), 3.92 (dd, J=9.1 , 6.1 , 1 H), 4.12 (t, J=QA, 1H), 4.44 (dd, J=11.7, 3.8, 1H), 4.49 (dd, J=11.7, 3.8, 1H), 4.88-4.94 (m, 1H), 5.16 (s, 2H), 6.93 (t, J=9.1 , 1 H), 7.08-7.12 (m, 1 H), 7.30-7.38 (m, 5H), 7.44 (dd, J=14.0, 2.6, 1 H).
Benzyl 4-(4-((S)-5-(aminomethyl)-2-oxo-oxazolidin-3-y!)-2-fluorophenyl) piperazine- 1-carboxylate (13). Compound 13 was obtained in 70% yield from 12 (4.4 g, 8:67 mmol), following the same procedure as for compound 6. After work-up, crude 13 was purified by flash chromatography using a gradient of 0-2-5-10% MeOH / CHCI3 as eluent. 1H NMR (400 MHz,
CDCI3) δ 1.33 (bs, 2H), 2.94-3.03 (m, 5H), 3.11 (dd, J=13.7, 4.1 , 1 H), 3.66-3.69 (m, 4H)1 3.82
(dd, J=8.6, 6.7, 1 H), 4.00 (t, J=8.7, 1 H)14.63-4.69 (m, 1 H), 5.16 (s, 2H), 6.91 (t, J=9.1 , 1 H)17.12- 7.15 (m, 1 H)1 7.30-7.38 (m, 5H)1 7.47 (dd, J=14.3, 2.6, 1 H).
Benzyl 4-(4-((S)-5-(acetylaminomethyl)-2-oxo-oxazolidin-3-yl)-2-fluorophenyl) piperazine-1-carboxylate (14). Compound 14 was obtained in 90% yield from 13 (5.3 g, 12.4 mmol), following the same procedure as for compound 7. After work-up, the compound was used without any further purification. 1H NMR (400 MHz, CDCI3) δ 2.02 (s, 3H), 3.01 (bs, 4H), 3.57-3.77 (m, 7H)14.01 (t, J=9.0, 1 H)14.73-4.79 (m, 1 H)1 5.16 (s, 2H)16.05 (t, J=6.2, 1H)16.91 (t, J=9.2, 1H), 7.05-7.08(m, 1 H)1 7.32-7.38 (m, 5H)1 7.44 (dd, J=14.2, 2.62, 1 H).
Λ/-[((S)-3-(3-fluoro-4-(piperazin-1-yl)phenyl]-2-oxo-oxazolidin-5-yl)methyl)acetamide (15). To a solution of 14 (748 mg, 1.59 mmol) in abs. ethanol (40 ml.) was added cyclohexene (1 ml.) and 10% Pd / C (400 mg). The mixture was refluxed for 2 h, when TLC indicated complete reaction. The reaction mixture was filtered through celite and concentrated to give 15 as an off-white solid (520 mg, 97%). The product was essentially pure, but could be purified by chromatography (90:10:1.5 CH2CI2:MeOH:conc. NH4OH). 1H NMR (400 MHz, CDCI3) 52.01 (s, 3H), 3.02 (d, J=Al, 8H), 3.57-3.76 (m, 3H), 4.01 (t, J=9.0, 1H), 4.73-4.79 (m, 1H), 6.29 (m, 1H)1 6.92 (t, J=9.1 , 1 H), 7.04-7.07(m, 1 H), 7.39-7.43 (m, 1 H).
Λ/-(((S)-3-(4-(4-(2-(benzyloxy)acetyl)piperazin-1-yl)-3-fluorophenyl)-2-oxooxazolidin- 5-yl)methyl)acetamide (16). To a solution of 15 (537 mg, 1.60 mmol) and triethylamine (0.22 mL, 3.53 mmol) in CH2CI2 (35 mL) at 0 0C was added benzyloxyacetyl chloride (0.30 ml_, 1.92 mmol). The mixture was stirred at 0 0C for 1 h, then 15 min at room temperature when TLC indicated complete reaction. The reaction mixture was washed with water (2 x 30 mL), and saturated sodium bicarbonate (2 x 30 mL), and dried over MgSO4. After chromatography (gradient elution 5-10% MeOH / CH2CI2) the product was obtained as a white foam (709 mg, 91%). 1H NMR (400 MHz, CDCI3) δ 2.02 (s, 3H), 2.98-3.14 (m, 4H), 3.56-3.86 (m, 7H), 4.02 (t, J=9.0, 1 H), 4.22 (S, 2H), 4.62 (s, 2H), 4.73-4.80 (m, 1H)1 6.02 (t, J=5.9,1 H), 6.96-7.10 (m, 2H), 7.28-7.40 (m, 5H), 7.45-7.53 (m, 1 H).
Λ/-(((S)-3-(3-fluoro-4-(4-(2-hydroxyacetyl)piperazin-1-yl)phenyl)-2-oxooxazoiidin-5- yl)methyl)acetamide (17, eperezolid). To a solution of 16 (709 mg, 1.46 mmol) in abs. ethanol (40 mL) was added cyclohexene (1 mL) and 10% Pd / C (250 mg). The mixture was refluxed for 15 h, when TLC indicated complete reaction. The reaction mixture was filtered through Celite™ and concentrated to give 17 (470 mg, 82% yield). The product was essentially pure, but could be purified by chromatography. 1H NMR (400 MHz, CDCI3) δ 2.02 (s, 3H), 3.06-3.10 (m, 4H), 3.45-3.50 (m, 2H), 3.58-3.77 (m, 3H), 3.85-3.87 (m, 2H), 4.02 (t, J=9.0, 1 H), 4.21 (s, 2H), 4.74- 4.80 (m, 1H), 6.09 (t, J=6.0, 1 H), 6.97 (t, J=QA , 1 H), 7.07-7.10 (m, 1 H), 7.46-7.50 (m, 1 H). LCMS : 96.1% (254 nm), 95.1% (220 nm), 94.5% (320 nm). MS : 395 (MH)+.
...................
EXAMPLE 8 (S)-N-[[3-[3-fluoro-4- 4-(hydro3ζyacetyl)-l-piperazinyl]-phenyl]-2- oxo-5-oxazoHdinyl]methylJ-acetamide sesquihydrate (VIII) To a stirred mixture of (S)-N-[[3-[3-fluoro-4-(l-piperazinyl)phenyl]-2-oxo-5- oxazoHdinyl]methyl]acetamide hydrochloride (EXAMPLE 7, 16.2 kg, 43.5 moles), tetrahydrofuran (205 kg) and triethylamine (10.1 kg, 100 moles) is added acetoxyacetyl chloride (6.5 kg, 47.8 moles) in tetrahydrofuran (11.1 kg) over 35 minutes keeping the temperature at 22-23°. After 40 minutes, at which time TLC and HPLC analysis indicated complete formation of the acetoxyacetamide intermediate, the mixture is concentrated under reduced pressure to 30 1, diluted with methanol (100 1) and concentrated to 30 1. To the residue is added methanol (25 1) and an aqueous solution of potassium carbonate (5.6 kg in 56 1). The resulting mixture is stirred 20 hr at 22-25° at which time TLC and HPLC analysis indicates the reaction is complete. The pH is adjusted to 7-7.5 with hydrochloric acid (4 N, 14.3 1). The mixture is stirred 18 hr at 15-22° then 3 hrs at 2-5°. The soHds are collected on a filter, washed with water (68 1) and dried at 20-25° with recycled nitrogen to give the desired product. The crude product is dissolved in water (225 1) at 60-70°, clarified through a 0.6 micron filter, diluted with water rinse (55 1) and stirred 17 hrs. at 15°. The solids are collected on a filter, washed with water at 15° and dried at 45° with recycled nitrogen to a water content of 0.33%. These soHds are dissolved in a solution of ethyl acetate (143 1), methanol (65 1) and water (1.95 1) at 60-65°. The solution is cooled to 15-25° and stirred 16 hrs for crystallization. The soHds are coUected on a filter, washed with ethyl acetate (75 1) and dried with 45° nitrogen to give the desired product. The product is recrystallized two more times from water (147 1 then 133 1) at 60-70°, clarified each time through a 0.6 micron filter and rinsed with water (40 1 and 30 1). The soHds are dried on the filter at 30° with recycled nitrogen to give, after deagglomeration through a mill, the title compound as the sesquihydrate (6.45% water), TLC (siHca gel; methanol/methylene chloride, 5/95) Rf = 0.45; [α]D = -20° (c = 1.0, ethanol).
pamidronate eperezolid
12-8-2000
|
BICYCLIC OXAZOLIDINONES AS ANTIBACTERIAL AGENT
| |
8-4-2000
|
ASSAYS FOR MODULATORS OF ELONGATION FACTOR P ACTIVITY
| |
3-22-2000
|
Method of treating psoriasis, arthritis and reducing the toxicity of cancer chemotherapy
| |
12-17-1999
|
MULTIVALENT MACROLIDE ANTIBIOTICS MULTIVALENT MACROLIDE ANTIBIOTICS MULTIVALENT MACROLIDE ANTIBIOTICS
|
8-4-2004
|
BICYCLIC HETEROCYCLIC SUBSTITUTED PHENYL OXAZOLIDINONE ANTIBACTERIALS, AND RELATED COMPOSITIONS AND METHODS
| |
9-12-2003
|
Bicyclic heterocyclic substituted phenyl oxazolidinone antibacterials, and related compositions and methods
| |
8-20-2003
|
Bicyclic heterocyclic substituted phenyl oxazolidinone antibacterials, and related compositions and methods
| |
4-9-2003
|
Compositions and methods for treating bacterial infections
| |
2-12-2003
|
Piperidinyloxy and pyrrolidinyloxy oxazolidinone antibacterials
| |
2-5-2003
|
Oxazolidinone tablet formulation
| |
7-3-2002
|
Bicyclic heterocyclic substituted phenyl oxazolidinone antibacterials, and related compositions and methods
| |
11-30-2001
|
Treatment of urinary tract infections with antibacterial oxazolidinones
| |
10-3-2001
|
N-substituted amidine and guanidine oxazolidinone antibacterials and methods of use thereof
| |
6-27-2001
|
Enhancement of oxazolidinone antibacterial agents activity by using arginine derivatives
|
8-15-2012
|
Oxazolidinone derivatives with cyclic amidoxime or cyclic amidrazone pharmaceutical compositions thereof
| |
10-20-2010
|
Oxazolidinone derivatives
| |
7-31-2009
|
NOVEL OXAZOLIDINONE DERIVATIVES
| |
6-20-2008
|
PREPARATION AND UTILITY OF SUBSTITUTED OXZOLIDINONES
| |
9-19-2007
|
Antibiotic conjugates
| |
3-31-2006
|
Antibiotic conjugates
| |
10-5-2005
|
Pyridoarylphenly oxazolidinone antibacterials, and related compositions and methods
| |
4-8-2005
|
Container for linezolid intravenous solution
| |
1-21-2005
|
Substituted isoxazoles and their use as antibiotics
| |
9-29-2004
|
Container for linezolid intravenous solution
|
6.........................
MRX 1
MRX I
MRX-I
1112968-42-9 cas no
C18 H15 F3 N4 O4
- 4(1H)-Pyridinone, 2,3-dihydro-1-[2,3,6-trifluoro-4-[(5S)-5-[(3-isoxazolylamino)methyl]-2-oxo-3-oxazolidinyl]phenyl]-
IN phase 1 FOR GRAM POSITIVE BACTERIA
MicuRx Pharmaceuticals (USA)
MicuRx Pharmaceuticals is developing two oxazolidinone compounds MRX-I and MRX-II. MRX-I is an oral oxazolidinone antibiotic that targets infections due to resistant Gram-positive bacteria, including MRSA and vancomycin-resistant enterococci (VRE). The company announced the completion of a double-blinded, placebo-controlled Phase 1 clinical study, and that the compound has been shown to be safe and well-tolerated at all doses tested with no evidence of myelosuppression.
In October 2012, the company announced the establishment of Shanghai MengKe Pharmaceuticals, a joint venture with Shanghai Zhangjiang Biomedical Industry Venture Capital formed to fund the development and commercialization of MRX-I for the Chinese market. MRX-II is currently under pre-clinical development [1,2].
MRX-I: A Potent and Safe Oxazolidinone Antibiotic
MRX-I is a next-generation oral oxazolidinone antibiotic for treating Gram-positivebacterial infections, including methicillin-resistant Staphylococcus aureus (MRSA) and vancomycin-resistant enterococci (VRE). In April 2012, MicuRx announced positive Phase I clinical results demonstrating that MRX-I is safe and well tolerated in human subjects, with no signs of myelosuppression, a major toxicity concern for most oxazolidinone agents, including linezolid.In preclinical studies, MRX-I cures in vivoinfections due to Gram-positive bacteria including MRSA and VRE effectively. In addition, MRX-I exhibits 2-fold improved activity against MRSA strains as compared to linezolid.
WO 2009020616 OR
http://www.google.fm/patents/US20090048305?cl=ja
Example 1 Compound of Structure

Scheme for the Compound of Example 1

Intermediate 17. 2,3,4,5-Tetrafluoronitrobenzene (1.17 g, 6.0 mmol) in N-methylpiperidone (NMP; 25 mL) was added dropwise with stirring to 4-piperidone hydrochloride (0.84 g, 6.2 mmol) and N,N-diisopropyl-N-ethylamine (DIEA; 2.45 mL, 14.0 mmol) in NMP (20 mL) at ca.-10 to −5° C. under nitrogen. The mixture was allowed to warm up to r.t. and stirred o.n. The mixture was taken into EtOAc (ca. 100 mL), washed with 2% aq. citric acid (2×50 mL), water (10×50 mL), brine, and dried (Na2SO4). Solvent was removed under vacuum, and the crude product was washed with hexanes (4×20 mL) and dried. Yellow crystals.1H NMR (400 MHz): 7.74 (m, 1H); 3.73 (t, J=6.0 Hz, 4H); 2.66 (t, J=6.0 Hz, 4H). MS (m/z): 275 [M+H].
Intermediate 18. Triethylamine (TEA; 5.6 mL, 43.87 mmol) was added to the Intermediate 17 (8.1 g, 29.56 mmol) in THF (120 mL) at 0° C., followed by triisopropylsilyl triflate (TIPSOTf; 10.7 g, 34.97 mmol). The mixture was allowed to warm up to r.t. over ca. 40 min, and stirred for another 2 h. Solvent was removed on a rotary evaporator. EtOAc (180 mL) was added, and the solution washed with 10% aq. NaHCO3 (40 mL), brine (60 mL) and dried (Na2SO4). Solvent was removed under vacuum and to afford the product as a red-brownish oil. This was directly used at the next step without purification.
Intermediate 19. Ceric ammonium nitrate (CAN, 19.0 g, 34.65 mmol) was added portionwise with stirring to a solution of the Intermediate 18 (12.4 g, 28.80 mmol) in dry DMF (100 mL) at 0° C. The reaction mixture was allowed to warm up to r.t. and stirred for another 4 h. Most of solvent was removed under vacuum. Water (ca. 75 mL) was added and the mixture was extracted with EtOAc (2×100 mL). The combined organic layers were washed with brine and dried (Na2SO4). Solvent was removed and the residue purified by column chromatography (gradient 20% to 30% EtOAc in petroleum ether). The product was obtained as a yellow solid. 1H NMR (400 MHz): 7.84 (m, 1H); 7.14 (m, 1H); 5.43 (d, J=8.2 Hz, 1H); 4.06 (t, J=7.2 Hz, 2H); 2.74 (t, J=7.2 Hz, 2H). MS (m/z): 273 [M+H].
Intermediate 20. NH4Cl (0.33 g, 6.2 mmol) in water (5 mL) was added to a hot solution of the Intermediate 19 (0.170 g, 0.62 mmol) in EtOH (10 mL). Iron powder (0.173 g, 3.1 mmol) was added portionwise with stirring, and the mixture at ca. 100-105° C. for 50 min. The solution was filtered, and the precipitate washed with EtOH (5×10 mL). EtOH was removed under vacuum, and residue distributed between EtOAc (ca. 50 mL) and water (10 mL). Aq. layer was washed with EtOAc (3×20 mL), and combined organic layers were washed with water (3×7 mL), brine, and dried (MgSO4). Solvent was removed under vacuum to afford the product as yellow crystals. 1H NMR (400 MHz): 7.03 (m, 1H); 6.36 (m, 1H); 5.19 (d, J=8.0 Hz, 1H); 4.12 (d, J=7.2 Hz, 2H); 3.80 (t, J=7.2 Hz, 2H); 2.66 (t, J=7.2 Hz, 2H). MS (m/z): 243 [M+H].
Intermediate 21.60% NaH in mineral oil (1.4 g, 36.0 mmol) was added portionwise with stirring to the Intermediate 20 (2.9 g, 11.94 mmol) in THF (20 mL) at 0° C. under Ar, and the mixture was stirred at this temperature for 30 min. Benzyl chloroformate (4.1 g, 24.03 mmol) was added dropwise with stirring. The reaction mixture was allowed to warm up to r.t. and stirred o.n. The reaction was carefully quenched with water (10 mL), and THF was removed under vacuum. The residue was taken in DCM (80 mL). Organic layer was washed with brine (50 mL) and dried (Na2SO4). Solvent was removed under vacuum, and the residue dissolved with MeOH (40 mL). Aq. NH3 (25 mL) was added with stirring, and the mixture was stirred at r.t. for 2 h. Solvent was removed under vacuum, and EtOAc (100 mL) was added. The organic layer was washed with brine and dried (Na2SO4). Solvent was removed under vacuum, and the residue purified by column chromatography (gradient 25% to 100% DCM/petroleum ether). White solid. 1H NMR (400 MHz): 7.95 (m, 1H); 7.41 (m, 6H); 7.07 (m, 2H); 5.28 (s, 2H); 3.88 (t, J=7.6 Hz, 2H); 2.69 (t, J=7.6 Hz, 2H). MS (m/z): 377 [M+H].
Compound of Example 1. 1.06M Lithium hexamethyldisilylamide (LHMDS; 3.0 mL, 3.18 mmol) in THF was added dropwise with stirring to a solution of the Intermediate 21 (1.0 g, 2.66 mmol) in THF (8.0 mL) at −78° C., and the mixture was stirred at this temperature for 30 min. (R)-Glycidyl butyrate (0.8 mL, 5.55 mmol) was added dropwise, and the mixture was allowed to warm up to r.t. and stirred o.n. The reaction was quenched with 10% aq. NH4Cl (15 mL), and THF was removed under vacuum. The residue was extracted with EtOAc (2×30 mL). Combined organic layers were washed with brine and dried (Na2SO4). Solvent was removed under vacuum. MeOH (5 mL) and 20% aqueous Cs2CO3 (5 mL) were added, and the mixture was stirred at r.t. for 20 min. The mixture was taken into EtOAc (50 mL), washed with water (2×15 mL), brine, and dried (Na2SO4). Solvent was removed under vacuum and the crude product was purified by column chromatography (2% methanol/DCM). Off-white solid. 1H NMR (400 MHz): 7.44 (m, 1H); 7.10 (d, J=7.6 Hz, 1H); 5.33 (d, J=8.0 Hz, 1H); 4.84 (m, 1H); 4.19 (m, 1H); 4.08 (m, 2H); 3.92 (t, J=7.4 Hz, 2H); 3.81 (dd, J=12.4, 3.2 Hz, 1H); 2.71 (t, J=7.4 Hz, 2H); 2.14 (br, 1H). MS (m/z): 343 [M+H].
......................................
WO2010091272A1
http://www.google.com/patents/WO2010091272A1?cl=en
Scheme 4 below.

Scheme 4. Example for synthesis of (isoxazole-3-yl)amino compounds of formula I.
a) Piperidin-4-one hydrochloride, DIEA, NMP, -5 0C to r.t; b) TMSOTf,
TEA, THF, 0 0C to r.t.; c) O-allyl-0' -methyl carbonate, Pd(OAc)2, DMSO, 2,3,4,5-tetrafluoronitrobenzene, 60 0C; d) Fe, NH4Cl, EtOH, 95 0C; e) isobutyl chloroformate, Py, DCM, 0 0C to r.t.; f) two steps: 1) (Λ)-glycidyl butyrate or chlorohydrine, Bu1OLi, THF, MeCN, 0-30 0C; 2) 10% aq. K2CO3; g) MsCl, TEA, THF, 0 0C; h) 3-[N-(/er/-butoxycarbonyl)amino]isoxazole, Bu1OK, DMF, 20-40 0C; i) aq. HCl, EtOH, EtOAc, 0 0C to r.t.
Select innovative steps pertaining to the particular utility of Scheme 4 for an efficient synthesis and production of the compounds of formula I (illustrated by structure 26 in the Scheme 4) are summarized in paragraphs (i-iv) below:
i) The novel efficient method for an installation of the dihydropyridone ring into an ortho-F compound of formula I provided herein involve the use of an alkoxide (e.g, methoxide) capture reagent (e.g., 2,3,4,5-tetrafluoronitrobenzene). The dihydropyri done-forming step for a transformation of the compounds 19 to compounds 20 performed in absence of the methoxide-capture reagent(s) is accompanied by formation of the hard-to-remove ort/zo-methoxy impurity (e.g., l-(2,6-difluoro-3-methoxy-4-nitrophenyl)-2,3-dihydropyridone) resulted from undesired substitution of ortho-F atom with MeOH, AIkOH, or anion thereof. This is a serious problem specific for the synthesis of ortho-F dihydropyridone compounds, arising from the unique reactivity of ortho-F substrates 19 and may not be encountered in synthesis of des-ortho-F compounds lacking the key ortho-F substitution. The methods disclosed herein involve the use of a methoxide-capture nitrobenzene additive to eliminate or minimize above methoxy-aryl by-product to allow for a high-yielding preparation and manufacture of precursors 19 and compounds of formula I, with a purity suitable for pharmaceutical applications (generally, better than 90-95%). Additional MeO-capture additives may include acylating, alkylating, or arylating agents (e.g., carboxylic acid anhydride or an active ester capable of methoxide acylation). Optionally, one or more alkoxide-capture reagent(s), or a combination thereof can be used.
ii) New practical method for the key oxazolidinone-forming step (from
22 to 23) provided herin involves the use of an alkali metal alkoxide (e.g., LiOBu- 1) instead of the conventionally used BuLi (as more generally described, e.g., in J. Med. Chem., 1988, vol. 41, pp. 3727-3735). The procedure provided herein thus eliminates the use of a highly flammable and unstable organometallic chemical. Moreover, the new processes provided herein also eliminates the need for costly cryogenic (-78 0C) conditions impractical for the industrial manufacture of the reagents 23 and of the compounds of formula I. [00111] iii) Novel process for the preparation of 5-[(isoxazole-3-yl)amino]methyl derivatives 25 that employs an alkali metal alkoxide ( e.g., KOBu-t) in place of previously used NaH (as more generally described, e.g., in International Patent Publication No. WO 00/21960, incorporated herein by reference in its entirety). This eliminates the use of an extremely flammable base and allows for an efficient preparation and manufacture of the precursors 25 and the compounds of formula I.
iv) New practical method for the synthesis of the compounds of formula I
(Ri = (isoxazole-3-yl)amino; structure 26 in Scheme 4) employing aq. HCl - organic solvent(s) system for deprotection of acid-cleavable protective groups (PG; e.g., PG = tert-butoxycarbonyl or Boc group). The method provided herein eliminates the use of highly toxic and expensive reagents conventionally employed for des-ortho-F 1-phenyldihydropyridone compounds (the method as described, for example, in International Patent Publication No. WO 2004/033449, advocating the use of trifluoroacetic acid and 1 ,2-dichloroethane Boc-deprotection system). The efficiency of the new deprotection method invented herein is particularly surprising in view of the fact that enamino ketones (such as dihydropyridones) are generally degradable by a strong aqueous acids, such as aq. HCl (as more generally described, e.g., by Katritzky et al. in J. Chem. Research, Miniprint, 1980, pp. 3337-3360).
.....................................
US8178683
https://www.google.com/patents/US8178683
Example 5 Compound of Structure
Scheme for Compound of Example 5
Intermediate 25.
Method A. A solution of tert-butyl isoxazol-3-ylcarbamate (187 mg, 1.00 mmol) in DMF (1 mL) was added dropwise with stirring to a suspension of NaH (60% in mineral oil, 48 mg, 1.20 mmol) in DMF (2 mL). The mixture was stirred under N2 for 15 min. at 35° C. The Intermediate 22 (357 mg, 0.85 mmol) in DMF (1 mL) was added, and the mixture was stirred at 50° C. for 1.5 h. The reaction mixture was taken into EtOAc (30 mL), washed with 10% aq. NH4Cl (2×15 mL), brine, and dried (Na2SO4). Solvent was removed under vacuum and the crude material was purified by column chromatography (2% MeOH/DCM) to afford the product as a light yellow solid.
Method B. A solution of tert-butyl isoxazol-3-ylcarbamate (694 mg, 3.8 mmol) in DMF (3 mL) was added dropwise with stirring to ButOK (439 mg, 3.8 mmol) in DMF (3 mL) at 0° C. The mixture was warmed up to r.t. and stirred for 30 min. The Intermediate 22 (1.34 g, 3.2 mmol) in DMF (6 mL) mL) was added, and the mixture was stirred at 35° C. for 2 h. The reaction was quenched with saturated aq. NH4Cl solution (10 mL), and isolation performed just as described above for Method A to afford the product as a light yellow solid. 1H NMR (400 MHz): 8.28 (s, 1H), 7.44 (m, 1H), 7.09 (d, J=7.6 Hz, 1H), 7.00 (s, 1H, 5.32 (d, J=7.6 Hz, 1H), 5.15 (m, 1H), 4.44 (m, 1H), 4.20 (m, 2H, 3.94 (m, 3H), 2.70 (t, J=7.4 Hz, 2H), 1.45 (s, 9H). MS (m/z): 509 [M+H].
Compound of Example 5
Method A. TFA (2.0 mL) was added dropwise to the solution of the Intermediate 25 (310 mg, 0.61 mmol) in 1,2-dichloroethane (DCE; 2 mL) at 0° C., and the solution was stirred at 0° C. for 30 min. Volatiles were removed under vacuum, and the residue taken into EtOAc (30 mL). The solution was washed with saturated NaHCO3 solution (2×15 mL), brine, and dried (Na2SO4). Solvent was removed under vacuum and the crude product was purified by column chromatography (3% MeOH/DCM). Light-yellow solid.
Method B. 4M HCl in THF (56 mL) was added dropwise to the Intermediate 25 (3.0 g, 5.9 mmol) at 0° C. Water (0.59 mL) was added, and the solution was stirred at r.t. for 2 h. Most of volatiles were removed under vacuum, the residue taken into water (30 mL) and sat. aq. NaHCO3 (15 mL), and pH adjusted to ca. 8. After stirring for 15 min, the mixture was extracted with EtOAc (3×60 mL). Combined organic layers were washed with brine (2×30 mL), and dried (Na2SO4). Solvent was removed under vacuum. The residue was re-dissolved in 2% MeOH in DCM (3 mL), and passed through a short pad of silica, eluting the product with 2% MeOH in DCM. Light-yellow solid. 1H NMR (400 MHz, DMSO-d6): 8.41 (d, J=1.6 Hz, 1H); 7.57 (m, 1H), 7.50 (d, J=8.0 Hz, 1H), 6.58 (t, J=5.8 Hz, 1H), 6.02 (d, J=1.6 Hz, 1H), 5.08 (d, J=8.0 Hz, 1H), 4.90 (m, 1H), 4.17 (t, J=8.6 Hz, 1H), 3.86 (m, 3H), 3.48 (t, J=5.6 Hz, 2H), 2.49 (m, overlapped with DMSO-d6, 2H). MS (m/z): 409 [M+H].
pick up int 22
from below
Example 3 Compound of Structure
Scheme for Compound of Example 3
Intermediate 22. Methylsulfonyl chloride (MsCl; 79 uL, 1.00 mmol) was added dropwise with stirring to the compound of Example 1 (290 mg, 0.85 mmol) and TEA (177 uL, 1.27 mmol, 1.50 equiv.) in DCM (5 mL) at ca. 0° C. The mixture was stirred for 20 min and allowed to warm up to r.t. The reaction mixture distributed between water and the DCM. Aq. layer was extracted with DCM (2×10 mL), and the combined organic layers washed with brine and dried (Na2SO4). Solvent was removed under vacuum to afford the product that was used for the next step without purification.
Intermediate 23. A mixture of the Intermediate 22 (567 mg, 1.35 mmol) and NaN3 (438 mg, 6.75 mmol) in DMF (5 mL) was stirred at 55° C. o.n. After cooling to r.t., water (15 mL) was added, and the reaction mixture was extracted with DCM (3×30 mL). Combined organic layers were washed with brine (30 ml) and dried (Na2SO4). Solvent was removed under vacuum to afford the product as a light yellow solid. This was used directly for the next step without further purification.
Compound of Example 3. A mixture of the Intermediate 23 (785 mg, 2.14 mmol) and bicyclo[2.2.1]hepta-2,5-diene (2.2 mL, 21.4 mmol) in 1,4-dioxane (22 mL) under N2 was heated at 100° C. for 3 h. Most of volatiles were removed under vacuum, and the residue was purified by column chromatography (1% MeOH/DCM). Thus isolated product was recrystallized from MeOH. White solid. 1H NMR (400 MHz): 7.83 (s, 2H), 7.05 (m, 2H), 5.30 (d, J=8 Hz, 1H), 5.16 (m, 1H), 4.83 (d, J=3.6 Hz, 2H), 4.33 (m, 1H), 4.06 (m, 1H), 3.91 (t, J=14.8 Hz, 2H), 2.69 (t, J=14.8 Hz, 2H). MS (m/z): 394 [M+H].
- MicuRx Pharmaceuticalsresistant Gram-positive bacteria, Inc. MicuRx and Shanghai Zhangjiang biomedical industry venture capital partner to develop next-generation antibiotic MRX-I for Chinese market. Available online: http://www.micurx.com/doc/10-24-12%20JV-FINAL.doc (accessed on 11 April 2013).
- MicuRx Pharmaceuticals. Discovery and development. Available online: http://www.micurx.com/d1.htm(accessed on 12 December 2012).
- CN 102206213
- CN 102485224
- CN 102485225
| US5668286 * | Pharmacia & Upjohn Company | Oxazolidinone derivatives and pharmaceutical compositions containing them | ||
| US6919329 * | Feb 24, 2003 | Jul 19, 2005 | Pharmacia & Upjohn Company | N-Aryl-2-oxazolidinone-5-carboxamides and their derivatives |
| US7105547 * | Oct 3, 2003 | Sep 12, 2006 | Pharmacia And Upjohn Company | Antimicrobial 1-aryl dihydropyridone compounds |
| US7141588 * | Aug 22, 2003 | Nov 28, 2006 | Pfizer, Inc. | N-aryl-2-oxazolidinone-5-carboxamides and their derivatives |
| WO2003006440A2 | Jul 12, 2002 | Jan 23, 2003 | Jackson B Hester Jr | Amide-containing compound having improved solubility and method of improving the solubility of an amide-containing compound |
| WO2003072553A1 * | Feb 24, 2003 | Sep 4, 2003 | Upjohn Co | N-aryl-2-oxazolidinone-5-carboxamides and their derivates and their use as antibacterials |
| WO2004033449A1 * | Oct 3, 2003 | Apr 22, 2004 | Mikhail Fedor Gordeev | Antimicrobial 1-aryl dihydropyridone compounds |
| WO2004059120A1 | Dec 16, 2003 | Jul 15, 2004 | Baker Hughes Inc | Anchor device to relieve tension from the rope socket prior to perforating a well |
| WO2004087697A1 | Mar 22, 2004 | Oct 14, 2004 | Christina Renee Harris | N-aryl-2-oxazolidinone-5-carboxamides derivatives with antibacterial activity |
| WO2005019213A1 | Aug 9, 2004 | Mar 3, 2005 | Robert Charles Gadwood | N-aryl-2-cyanooxazolidinones and their derivatives |
| WO2005113520A1 | May 9, 2005 | Dec 1, 2005 | Michael Robert Barbachyn | Substituted 2,3,5-trifluorphenyl oxazolidinones for use as antibacterial agents |
| WO2006038100A1 | Oct 6, 2005 | Apr 13, 2006 | Ranbaxy Lab Ltd | Oxazolidinone derivatives as antimicrobials |
| WO2007000644A1 | Jun 20, 2006 | Jan 4, 2007 | Pharmacia & Upjohn Co Llc | Homomorpholine oxazolidinones as antibacterial agents |
| WO2007004049A1 | Jun 26, 2006 | Jan 11, 2007 | Pharmacia & Upjohn Co Llc | Oxazolidinones containing azetidine as antibacterial agents |
.................................
7...................
LCB01-0371
LCB01-0371
3-[3-Fluoro-4-(1-methyl-1,4,5,6-tetrahydro-1,2,4-triazin-4-yl)phenyl]-5(R)-(hydroxymethyl)oxazolidin-2-one
LegoChem Biosciences (South Korea)
Phase I, Gram-positive
308.3082
C14 H17 F N4 O3
LCB01-0371 is being developed by LegoChem Bio. This new oxazolidinone has improved activity against Gram-positive pathogens and has good pharmacokinetic profiles in animals [103].
The compound is under Phase I clinical development to assess the safety and tolerability of the compound. The company is currently recruiting participants to be part of the trial [103,104].
read
- [PDF]
7. 레고켐(임상).cdr
www.kddf.org/common/file/?idx=1669&fname=7._레고켐(임상)...
New oxazolidinone LCB01-0371 for MRSA and VRE infection. Young Lag Cho. Company : LegoChem Biosciences, Inc. Website : http://www.legochembio.com.
LCB01-0371 is a new oxazolidinone with cyclic amidrazone. In vitro activity of LCB01-0371 against 624 clinical isolates was evaluated and compared with those of linezolid, vancomycin, and other antibiotics. LCB01-0371 showed good activity against Gram-positive pathogens. In vivo activity of LCB01-0371 against systemic infections in mice was also evaluated. LCB01-0371 was more active than linezolid against these systemic infections. LCB01-0371 showed bacteriostatic activity against Staphylococcus aureus.
As examples of oxazolidinone compounds including an oxazolidinone ring, 3-phenyl-2-oxazolidinone derivatives having one or two substituent(s) are described in US Patent Nos. 4,948,801, 4,461,773, 4,340,606, 4,476,136, 4,250,318 and 4,128,654, and 3-[(mono-substituted)phenyl]-2-oxazolidinone derivatives represented by Chemical Formula A are described in EP 0312000, J. Med. Chem.32, 1673(1989), J. Med. Chem. 33, 2569 (1990), Tetrahedron Lett. 45,123(1989), and the like.
[Chemical Formula A]

And, oxazolidinone derivatives represented by Chemical Formula B and Chemical Formula C were synthesized by Pharmacia & Upjohn (WO 93/23384, WO 95/14684 and WO 95/07271). The compound of Chemical Formula B, "linezolid", is the first oxazolidinone antibiotic and is marketed under the trade name "zyvox" for oral administration and injection, approved by the U.S. Food and Drug Administration (FDA). However, most of synthetic oxazolidinone compounds are associated with some limitations, such as toxicity, low in vivo efficacy and low solubility. As for linezolid, solubility in water is only about 3 mg/mL, which causes its use as injection limited.
[Chemical Formula B]

[Chemical Formula C]

WO 93/09103 discloses phenyl oxazolidinone derivatives having a heterocyclic ring, including pyridine, thiazole, indole, oxazole, quinol, etc., at the 4-position of the phenyl group. But, the substituents of the heterocyclic ring are merely simple alkyl or amino group, and the activities are not so excellent.
In order to solve these problems, WO 01/94342 discloses phenyloxazolidinone derivatives having various pyridine or phenyl derivatives at the 4-position of the phenyl group. The synthetic compounds have wide antibacterial spectrum and excellent antibacterial activity. Although the oxazolidinone compounds having various pyridine derivatives at the 4-position of the phenyl group of oxazolidinone have wider antibacterial spectrum and excellent antibacterial activity as compared to linezolid, most of them have aqueous solubility of 30 ㎍/mL or less, and thus have limitation in preparing injections.
TR-700 and TR-701, represented by Chemical Formula D, are developed by Dong-A Pharmaceutical and recently licensed to Trius Therapeutics. TR-701 is a prodrug of TR-700 and it is in the phase II clinical trial. TR-701 solves the solubility problem via formation of prodrug from TR-700, exhibits an antibacterial activity superior to that of linezolid. However, the compound shows higher toxicities (cytotoxicity, MAO profile, myelosuppression, etc.) than linezolid, and, thus, is expected to have many limitations.
[Chemical Formula D]

As described above, a compound having superior antibacterial activity, satisfactory solubility and lower toxicity is yet to be found.
The inventors of the present invention have synthesized novel oxazolidinone derivatives in order to develop antibiotics having superior antibacterial activity as compared to existing antibiotics and having higher solubility for easier preparation into oral administration and injection formulations. The novel oxazolidinone derivatives according to the present invention have been confirmed to have superior antibacterial activity and significantly improved antibacterial spectrum.
Especially, the cyclic amidoxime or cyclic amidrazone compound presented by the present invention has not been studied before. Whereas acyclic amidoxime or amidrazone is relatively well known, the cyclic amidoxime or cyclic amidrazone compound like those disclosed in the present invention is hardly known. Introduction of the cyclic form results in remarkably improved absorptivity and allows the formation of a salt having an adequate basicity, thereby greatly increasing solubility in water. The increased solubility in water makes it possible to prepare injections without using a prodrug and with little toxicity.
watch outfor synthesis...will be updated
WO 2010036000
http://www.google.com/patents/WO2010036000A2?cl=en
[Scheme 1]

[Scheme 2]


[Scheme 3]

[Scheme 4]


[Scheme 5]


*[Scheme 6]


http://www.google.com/patents/WO2010036000A2?cl=en
[Example 94] Preparation of Compound 94
 Compound 93 (150 mg, 0.51 mmol) was dissolved in methanol (5 mL), formaldehyde (37% aqueous solution, 0.21 mL, 2.55 mmol) and stirred for 1 hour at room temperature after adding acetic acid (0.03 mL, 0.51 mmol) and NaBH3CN (48 mg, 0.77 mmol). The solution was distilled under reduced pressure, dissolved in dichloromethane (100 mL), sequentially washed with saturated aqueous sodium bicarbonate solution and saturated aqueous sodium chloride solution (brine), dried with anhydrous sodium sulfate, concentrated under reduced pressure, and separated by column chromatography to obtain Compound 94(71 mg, 0.23 mmol, 45%).
Compound 93 (150 mg, 0.51 mmol) was dissolved in methanol (5 mL), formaldehyde (37% aqueous solution, 0.21 mL, 2.55 mmol) and stirred for 1 hour at room temperature after adding acetic acid (0.03 mL, 0.51 mmol) and NaBH3CN (48 mg, 0.77 mmol). The solution was distilled under reduced pressure, dissolved in dichloromethane (100 mL), sequentially washed with saturated aqueous sodium bicarbonate solution and saturated aqueous sodium chloride solution (brine), dried with anhydrous sodium sulfate, concentrated under reduced pressure, and separated by column chromatography to obtain Compound 94(71 mg, 0.23 mmol, 45%).
1H NMR (600 MHz, DMSO-d6) δ= 7.59 (dd, J 1 = 13.8 Hz, J 2 = 2.4 Hz, 1H), 7.33-7.30 (m, 2H), 6.84 (s, 1H), 5.23 (t, J = 5.4 Hz, 1H), 4.70 (m, 1H), 4.07 (t, J = 9.0 Hz, 1H), 3.82 (m, 1H), 3.71 (t, J = 4.8 Hz, 2H), 3.69-3.54 (m, 2H), 2.87 (t, J = 4.8 Hz, 2H), 2.61 (s, 3H).
LCMS: 309 (M + H+) for C14H17-FN4O3.
[Example 93] Preparation of Compound 93
 Compound 93 (190 mg, 0.65 mmol, 74%) was obtained from Compound 92 as in Example 2.
Compound 93 (190 mg, 0.65 mmol, 74%) was obtained from Compound 92 as in Example 2.
1H NMR (600 MHz, DMSO-d6) δ= 7.73 (dd, J 1 = 13.8 Hz, J 2 = 2.4 Hz, 1H), 7.60 (t, J = 9 Hz, 1H), 7.45 (dd, J 1 = 9.0 Hz, J2 = 2.4 Hz, 1H), 4.75 (m, 1H), 4.11 (t, J = 9.0 Hz, 1H), 3.88 (m, 1H), 3.78 (t, J = 4.8 Hz, 2H), 3.70-3.55 (m, 2H), 3.36 (t, J =4.8 Hz, 2H).
LCMS: 295 (M + H+) for C13H15-FN4O3.
[Example 92] Preparation of Compound 92
 Compound 92 (240 mg, 0.75 mmol, 32%) was obtained from Compound XXIII as in Preparation Example 10.
Compound 92 (240 mg, 0.75 mmol, 32%) was obtained from Compound XXIII as in Preparation Example 10.
1H NMR (600 MHz, CDCl3) δ= 8.55 (s, 1H), 7.61 (dd, J 1 = 13 Hz, J 2 = 2.4 Hz, 1H), 7.25 (dd, J 1 = 9.0 Hz, J 2 = 2.7 Hz, 1H), 7.14 (t, J = 8.4 Hz, 1H), 6.90 (s, 1H), 4.79 (m, 1H), 4.04-3.99 (m, 5H), 3.79-3.73 (m, 3H), 2.58 (br, s, 1H).
LCMS: 323 (M + H+) for C14H15-FN4O4.
[Preparation Example 17] Preparation of Compound XXIII
 Compound V (26 g, 0.053 mol) was dissolved in dichloromethane (180 mL) and stirred for 10 minutes after slowly adding diisopropylethylamine (DIPEA, 13 mL, 0.079 mol) and benzoyl chloride (Bz-Cl, 7.4 mL, 0.064 mol) sequentially dropwise at 0℃. After heating to room temperature, followed by adding a small amount of DMAP, the solution was stirred for 2 hours. The solution was concentrated under reduced pressure, dissolved in ethyl acetate, sequentially washed with saturated aqueous sodium bicarbonate solution and saturated aqueous sodium chloride solution (brine), dried with anhydrous sodium sulfate, and concentrated under reduced pressure to quantitatively obtain Compound XXII (31 g, 0.053 mol), which was treated with hydrochloric acid as in Preparation Example 9 to quantitatively obtain Compound XXIII.
Compound V (26 g, 0.053 mol) was dissolved in dichloromethane (180 mL) and stirred for 10 minutes after slowly adding diisopropylethylamine (DIPEA, 13 mL, 0.079 mol) and benzoyl chloride (Bz-Cl, 7.4 mL, 0.064 mol) sequentially dropwise at 0℃. After heating to room temperature, followed by adding a small amount of DMAP, the solution was stirred for 2 hours. The solution was concentrated under reduced pressure, dissolved in ethyl acetate, sequentially washed with saturated aqueous sodium bicarbonate solution and saturated aqueous sodium chloride solution (brine), dried with anhydrous sodium sulfate, and concentrated under reduced pressure to quantitatively obtain Compound XXII (31 g, 0.053 mol), which was treated with hydrochloric acid as in Preparation Example 9 to quantitatively obtain Compound XXIII.
[Preparation Example 5] Preparation of Compound V
 Compound IV (116 g, 0.22 mol) was dissolved in THF (400 mL) and stirred for 20 minutes after slowly adding n-butyllithium (2.5 M solution in n-hexane, 90 mL, 0.23 mol) dropwise at -78℃. After adding (R)-glycidyl butyrate (31.5 mL, 0.23 mol), followed by stirring for 3 hours while slowly heating to room temperature, the solution was adjusted to pH ~6 with aqueous ammonium chloride solution, and concentrated under reduced pressure. The concentrate was dissolved in 80% ethyl acetate/hexane solution, sequentially washed with water and saturated aqueous sodium chloride solution (brine), dried with anhydrous sodium sulfate, and concentrated under reduced pressure. The concentrate was separated by column chromatography using 40% ethyl acetate/hexane solution to obtain Compound V (45 g, 0.093 mol, 42%) as a colorless oil.
Compound IV (116 g, 0.22 mol) was dissolved in THF (400 mL) and stirred for 20 minutes after slowly adding n-butyllithium (2.5 M solution in n-hexane, 90 mL, 0.23 mol) dropwise at -78℃. After adding (R)-glycidyl butyrate (31.5 mL, 0.23 mol), followed by stirring for 3 hours while slowly heating to room temperature, the solution was adjusted to pH ~6 with aqueous ammonium chloride solution, and concentrated under reduced pressure. The concentrate was dissolved in 80% ethyl acetate/hexane solution, sequentially washed with water and saturated aqueous sodium chloride solution (brine), dried with anhydrous sodium sulfate, and concentrated under reduced pressure. The concentrate was separated by column chromatography using 40% ethyl acetate/hexane solution to obtain Compound V (45 g, 0.093 mol, 42%) as a colorless oil.
1H NMR (600 MHz, CDCl3) δ 7.50-7.48 (m, 1H), 7.30-7.28 (m, 1H), 7.17-7.16 (m, 1H), 4.74-4.70 (m, 1H), 4.03-4.02 (m, 1H), 3.98 (m, 2H), 3.75 (m, 3H), 3.65 (m, 2H), 1.51 (s, 3H), 1.36 (s, 6H), 0.85 (s, 9H), 0.02 (s, 6H).
[Preparation Example 1] Preparation of Compound I
 After dissolving 3,4-difluoronitrobenzene (158 g, 0.99 mol) in acetonitrile (800 mL) and adding ethanolamine (117 g, 1.9 mol), the mixture was stirred for 4 hours under reflux. The reaction solution was cooled to room temperature, concentrated under reduced pressure, triturated with diethyl ether, and filtered to obtain yellow Compound I (199 g, 0.99 mol, 100%).
After dissolving 3,4-difluoronitrobenzene (158 g, 0.99 mol) in acetonitrile (800 mL) and adding ethanolamine (117 g, 1.9 mol), the mixture was stirred for 4 hours under reflux. The reaction solution was cooled to room temperature, concentrated under reduced pressure, triturated with diethyl ether, and filtered to obtain yellow Compound I (199 g, 0.99 mol, 100%).
1H NMR (400 MHz, chloroform-d1) δ 7.97 (d, 1H, J = 8.8 Hz), 7.87 (dd, 1H, J 1 = 11.6 Hz, J 2 = 2.4 Hz), 6.65 (t, 1H, J = 8.8 Hz), 5.10-4.87 (bs, 1H), 3.97-3.83 (m, 2H), 3.43-3.37 (m, 2H).
[Preparation Example 2] Preparation of Compound II
 Compound I (100 g, 0.5 mol), t-butyldimethylsilyl chloride (TBS-Cl, 97 g, 0.65 mol) and imidazole (51 g, 0.75 mol) were dissolved in dichloromethane (700 mL) at 0℃ and stirred overnight after slowly heating to room temperature. The reaction solution was concentrated under reduced pressure, dissolved in ethyl acetate and washed with 0.5 N HCl, washed sequentially with saturated aqueous sodium bicarbonate solution and saturated aqueous sodium chloride solution (brine), dried with anhydrous sodium sulfate, and concentrated under reduced pressure to quantitatively obtain a compound with a tbs group attached to alcohol. This compound was dissolved in THF (500 mL) and 1.2 equivalents of Boc2O and 0.1 equivalent of 4-dimethylaminopyridine (DMAP) were added. After stirring for 3 hours at room temperature, ammonia water (30 mL) was added. After stirring further for 20 minutes, the solution was concentrated under reduced pressure. The concentrate was dissolved again in ethyl acetate, sequentially washed with 0.5 N HCl, saturated aqueous sodium bicarbonate solution and saturated aqueous sodium chloride solution (brine), dried with anhydrous sodium sulfate, and concentrated under reduced pressure to quantitatively obtain Compound II.
Compound I (100 g, 0.5 mol), t-butyldimethylsilyl chloride (TBS-Cl, 97 g, 0.65 mol) and imidazole (51 g, 0.75 mol) were dissolved in dichloromethane (700 mL) at 0℃ and stirred overnight after slowly heating to room temperature. The reaction solution was concentrated under reduced pressure, dissolved in ethyl acetate and washed with 0.5 N HCl, washed sequentially with saturated aqueous sodium bicarbonate solution and saturated aqueous sodium chloride solution (brine), dried with anhydrous sodium sulfate, and concentrated under reduced pressure to quantitatively obtain a compound with a tbs group attached to alcohol. This compound was dissolved in THF (500 mL) and 1.2 equivalents of Boc2O and 0.1 equivalent of 4-dimethylaminopyridine (DMAP) were added. After stirring for 3 hours at room temperature, ammonia water (30 mL) was added. After stirring further for 20 minutes, the solution was concentrated under reduced pressure. The concentrate was dissolved again in ethyl acetate, sequentially washed with 0.5 N HCl, saturated aqueous sodium bicarbonate solution and saturated aqueous sodium chloride solution (brine), dried with anhydrous sodium sulfate, and concentrated under reduced pressure to quantitatively obtain Compound II.
1H NMR (600 MHz, chloroform-d1) δ 8.06-7.98 (m, 1H), 7.95 (dd, 1H, J 1 = 10.2 Hz, J 2 = 2.4 Hz), 7.57 (t, 1H, J = 7.8 Hz), 3.80 (t, 2H, J = 5.4 Hz), 3.73 (t, 2H, J = 4.8 Hz), 1.42 (s, 9H), 0.81 (s, 9H), 0.01 (s, 6H).
[Preparation Example 3] Preparation of Compound III
 Compound II (92 g, 0.22 mol) was dissolved in methanol (600 mL) and stirred for 4 hours under hydrogen balloon after adding Pd/C (6 g). The reaction mixture was filtered using celite and concentrated under reduced pressure to quantitatively obtain Compound III (86 g) as a colorless oil.
Compound II (92 g, 0.22 mol) was dissolved in methanol (600 mL) and stirred for 4 hours under hydrogen balloon after adding Pd/C (6 g). The reaction mixture was filtered using celite and concentrated under reduced pressure to quantitatively obtain Compound III (86 g) as a colorless oil.
1H NMR (400 MHz, chloroform-d1) δ 6.99 (t, 1H, J = 12.0 Hz), 6.44-6.30 (m, 2H), 3.81-3.63 (m, 4H), 3.63-3.52 (m, 2H), 1.50 (s, 3H), 1.35 (s, 6H), 0.86 (s, 9H), 0.03 (s, 6H).
[Preparation Example 4] Preparation of Compound IV
 Compound III (86 g, 0.22 mol) was dissolved in dichloromethane (300 mL). After adding aqueous 1 N NaOH solution (300 mL), benzyl chloroformate (Cbz-Cl, 38 mL, 0.27 mol) was slowly added dropwise while stirring. After stirring for 1 hour at room temperature, the organic layer was separated, washed twice with water, dried with anhydrous sodium sulfate, and concentrated under reduced pressure to quantitatively obtain Compound IV (116 g) as a yellow oil.
Compound III (86 g, 0.22 mol) was dissolved in dichloromethane (300 mL). After adding aqueous 1 N NaOH solution (300 mL), benzyl chloroformate (Cbz-Cl, 38 mL, 0.27 mol) was slowly added dropwise while stirring. After stirring for 1 hour at room temperature, the organic layer was separated, washed twice with water, dried with anhydrous sodium sulfate, and concentrated under reduced pressure to quantitatively obtain Compound IV (116 g) as a yellow oil.
1H NMR (600 MHz, chloroform-d1) δ 7.44-7.32 (m, 6H), 7.18 (t, 1H, J = 8.1 Hz), 6.96 (d, 1H, J = 8.4 Hz), 6.84-6.66 (bs, 1H), 5.20 (s, 2H), 3.82-3.63 (m, 2H), 3.63-3.58 (m, 2H), 1.51 (s, 3H), 1.35 (s, 6H), 0.86 (s, 9H), 0.02 (s, 6H).
[Example 94] Preparation of Compound 94

1H NMR (600 MHz, DMSO-d6) δ= 7.59 (dd, J 1 = 13.8 Hz, J 2 = 2.4 Hz, 1H), 7.33-7.30 (m, 2H), 6.84 (s, 1H), 5.23 (t, J = 5.4 Hz, 1H), 4.70 (m, 1H), 4.07 (t, J = 9.0 Hz, 1H), 3.82 (m, 1H), 3.71 (t, J = 4.8 Hz, 2H), 3.69-3.54 (m, 2H), 2.87 (t, J = 4.8 Hz, 2H), 2.61 (s, 3H).
LCMS: 309 (M + H+) for C14H17-FN4O3.
[Example 93] Preparation of Compound 93

1H NMR (600 MHz, DMSO-d6) δ= 7.73 (dd, J 1 = 13.8 Hz, J 2 = 2.4 Hz, 1H), 7.60 (t, J = 9 Hz, 1H), 7.45 (dd, J 1 = 9.0 Hz, J2 = 2.4 Hz, 1H), 4.75 (m, 1H), 4.11 (t, J = 9.0 Hz, 1H), 3.88 (m, 1H), 3.78 (t, J = 4.8 Hz, 2H), 3.70-3.55 (m, 2H), 3.36 (t, J =4.8 Hz, 2H).
LCMS: 295 (M + H+) for C13H15-FN4O3.
[Example 92] Preparation of Compound 92

1H NMR (600 MHz, CDCl3) δ= 8.55 (s, 1H), 7.61 (dd, J 1 = 13 Hz, J 2 = 2.4 Hz, 1H), 7.25 (dd, J 1 = 9.0 Hz, J 2 = 2.7 Hz, 1H), 7.14 (t, J = 8.4 Hz, 1H), 6.90 (s, 1H), 4.79 (m, 1H), 4.04-3.99 (m, 5H), 3.79-3.73 (m, 3H), 2.58 (br, s, 1H).
LCMS: 323 (M + H+) for C14H15-FN4O4.
[Preparation Example 17] Preparation of Compound XXIII

[Preparation Example 5] Preparation of Compound V

1H NMR (600 MHz, CDCl3) δ 7.50-7.48 (m, 1H), 7.30-7.28 (m, 1H), 7.17-7.16 (m, 1H), 4.74-4.70 (m, 1H), 4.03-4.02 (m, 1H), 3.98 (m, 2H), 3.75 (m, 3H), 3.65 (m, 2H), 1.51 (s, 3H), 1.36 (s, 6H), 0.85 (s, 9H), 0.02 (s, 6H).
[Preparation Example 1] Preparation of Compound I

1H NMR (400 MHz, chloroform-d1) δ 7.97 (d, 1H, J = 8.8 Hz), 7.87 (dd, 1H, J 1 = 11.6 Hz, J 2 = 2.4 Hz), 6.65 (t, 1H, J = 8.8 Hz), 5.10-4.87 (bs, 1H), 3.97-3.83 (m, 2H), 3.43-3.37 (m, 2H).
[Preparation Example 2] Preparation of Compound II

1H NMR (600 MHz, chloroform-d1) δ 8.06-7.98 (m, 1H), 7.95 (dd, 1H, J 1 = 10.2 Hz, J 2 = 2.4 Hz), 7.57 (t, 1H, J = 7.8 Hz), 3.80 (t, 2H, J = 5.4 Hz), 3.73 (t, 2H, J = 4.8 Hz), 1.42 (s, 9H), 0.81 (s, 9H), 0.01 (s, 6H).
[Preparation Example 3] Preparation of Compound III

1H NMR (400 MHz, chloroform-d1) δ 6.99 (t, 1H, J = 12.0 Hz), 6.44-6.30 (m, 2H), 3.81-3.63 (m, 4H), 3.63-3.52 (m, 2H), 1.50 (s, 3H), 1.35 (s, 6H), 0.86 (s, 9H), 0.03 (s, 6H).
[Preparation Example 4] Preparation of Compound IV

1H NMR (600 MHz, chloroform-d1) δ 7.44-7.32 (m, 6H), 7.18 (t, 1H, J = 8.1 Hz), 6.96 (d, 1H, J = 8.4 Hz), 6.84-6.66 (bs, 1H), 5.20 (s, 2H), 3.82-3.63 (m, 2H), 3.63-3.58 (m, 2H), 1.51 (s, 3H), 1.35 (s, 6H), 0.86 (s, 9H), 0.02 (s, 6H).
- 103 Jeong, J.-W.; Jung, S.-J.; Lee, H.-H.; Kim, Y.-Z.; Park, T.-K.; Cho, Y.-L.; Chae, S.-E.; Baek, S.-Y.; Woo, S.-H.; Lee, H.-S.; et al. In vitro and In vivo activities of LCB01–0371, a new oxazolidinone. Antimicrob. Agents Chemother. 2010, 54, 5359–5362, doi:10.1128/AAC.00723-10.
- 104 LegoChem Biosciences. Multiple ascendoing dose study for LCB01–0371. Available online: http://www.clinicaltrials.gov/ct2/show/NCT01842516 (accessed on 15 August 2013).
- http://clinicaltrials.gov/ct2/show/NCT01842516
- http://www.pubfacts.com/author/Yong+Zu+Kim
- New oxazolidinones with cyclic amidrazone (I): Structure activity relationship of cyclic amidrazone antibiotics
49th Intersci Conf Antimicrob Agents Chemother (ICAAC) (September 12-15, San Francisco) 2009, Abst F1-1508 - [PDF]
7. 레고켐(임상).cdr
www.kddf.org/common/file/?idx=1669&fname=7._레고켐(임상)...New oxazolidinone LCB01-0371 for MRSA and VRE infection. Young Lag Cho. Company : LegoChem Biosciences, Inc. Website : http://www.legochembio.com.
| KR100674096B1 * | Title not available | |||
| KR100713170B1 * | Title not available | |||
| KR20040035207A * | Title not available | |||
| US7157456 * | Dec 11, 2000 | Jan 2, 2007 | Bayer Healthcare Ag | Substituted oxazolidinones and their use in the field of blood coagulation |
| WO2011111971A2 * | Mar 8, 2011 | Sep 15, 2011 | Legochem Biosciences,Inc. | Method for preparing (r)-3-(3-fluoro-4-(1-methyl-5,6-dihydro-1,2,4-triazin-4(1h)-yl)phenyl)-5-(substituted methyl)oxazolidin-2-one derivatives |
| WO2012121424A1 * | Mar 4, 2011 | Sep 13, 2012 | (주)레고켐바이오사이언스 | Novel oxazolidinone derivative having cyclic amidrazone group and pharmaceutical composition containing same |
...............................................................
8 RANBEZOLID
Ranbezolid
392659-39-1 hydrochloride
392659-38-0 (free base)
N-{[(5S)-3-(3-Fluoro-4-{4-[(5-nitro-2-furyl)methyl]-1-piperazinyl}phenyl)-2-oxo-1,3-oxazolidin-5-yl]methyl}acetamide
(S)-N-[[3-fluoro-4-[N-1[4-{2-furyl-(5-nitro)methyl}]piperazinyl]-phenyl]-2-oxo-5-oxazolidinyl]-methyl]acetamide
AC1LAX1P, RBx7644 (*Hydrochloride*),RBx-7644
Molecular Formula: C21H24FN5O6 Molecular Weight: 461.443563
Ranbaxy Lab Ltd ORIGINATOR
Ranbezolid is a novel oxazolidinone antibacterial. It competitively inhibits monoamine oxidase-A (MAO-A).[1]
Infections due to Gram-positive bacteria such as methicillin-resistant Staphylococcus aureus (MRSA), vancomycin-resistant Enterococcus faecium (VRE), and penicillin-resistant Streptococcus pneumoniae(PRSP) are the leading cause of morbidity and mortality in hospital settings and community today. Oxazolidinones are a new class of totally synthetic antibacterial agents active against Gram-positive infections. Linezolid (Zyvox™, Pharmacia/Pfizer, is a drug in this class, approved in the United States and Europe for treatment of Gram-positive nosocomial and community-acquired pneumoniae and skin infections. Oxazolidinones inhibit the bacterial protein synthesis prior to the chain initiation step, by binding to the 23S rRNA of 50S ribosomal subunit, and interfering with the initiator fMet–tRNA binding to the P-site of the ribosomal peptidyltransferase centre
Ranbezolid hydrochloride, RBx-7644
9-23-2005
|
Plymorphic forms of phenyl oxazolidinone derivatives
|
The title compound is prepared by reductive alkylation of the known piperazinyl oxazolidinone derivative (I) with 5-nitro-2-furfural (II) in the presence of NaBH(OAc)3, followed by conversion to the corresponding hydrochloride salt.
EP 1303511; US 2002103186; WO 0206278; WO 0307870; WO 0308389
................
synthesis
The antibacterial activity of RBx-7644 is due to the 5(S)-acetamidomethyl configuration at the oxazolidinone ring, and thus, asymmetric synthesis of only the 5(S)-enantiomer was desirable: 3,4-Difluoronitrobenzene (I) is condensed with piperazine in acetonitrile to give 4-(2-fluoro-4-nitrophenyl)-piperazine (II) as a light yellow compound. Compound (II) is dissolved in dichloromethane and triethylamine, followed by the addition of Boc-anhydride, to provide compound (III). 4-(tert-Butoxycarbonyl)-1-(2-fluoro-4-nitrophenyl)piperazine (III), upon hydrogenation with H2 over Pd/C in methanol at 50 psi, yields 4-(tert-butoxycarbonyl)-1-(2-fluoro-4-aminophenyl)piperazine (IV) as a dark solid. Compound (IV) reacts with benzylchloroformate in dry THF in the presence of solid sodium bicarbonate to afford the desired compound (V). 4-(tert-Butoxycarbonyl)-1-[2-fluoro-4-(benzyloxycarbonylamino)phenyl]piperazine (V), upon treatment with n-BuLi and (R)-glycidyl butyrate at -78 癈, gives the desired (R)-(-)-3-[3-fluoro-4-[4-(tert-butoxycarbonyl)piperazin-1-yl]phenyl]-5-(hydroxymethyl)-2-oxazolidinone (VI). The hydroxymethyl compound (VI) is treated with methanesulfonyl chloride in dichloromethane in the presence of triethylamine to give (R)-(-)-3-[3-fluoro-4-[4-(tert-butoxycarbonyl)piperazin-1-yl]phenyl]-5-(methylsulfonyloxymethyl)-2-oxazolidinone (VII). The sulfonyl derivative (VII) is treated with sodium azide in dimethylformamide to provide the azide (VIII) as a white solid. (R)-(-)-3-[3-Fluoro-4-[4-(tert-butoxycarbonyl)piperazin-1-yl)phenyl]-5-(azidomethyl)-2-oxazolidinone (VIII), upon hydrogenation with H2 over Pd/C at 45 psi, gives (S)-(-)-3-[3-fluoro-4-[4-(tert-butoxycarbonyl)-piperazin-1-yl]phenyl]-5-(aminomethyl)-2-oxazolidinone (IX). The aminomethyl compound (IX), upon treatment with acetic anhydride in dichloromethane in the presence of triethylamine, affords the acetamide derivative (X). The acetamidomethyl-oxazolidinone derivative (X), upon treatment with trifluoroacetic acid, gives (S)-(-)-3-[3-fluoro-4-(1-piperazinyl)phenyl]-5-(acetamidomethyl)-2-oxazolidinone, which, without isolation, is treated with 5-nitro-2-furaldehyde in the presence of sodium triacetoxy borohydride to provide compound (XI). Compound (XI), upon treatment with ethanolic HCl, affords RBx-7644 as a light yellow crystalline solid.
......................
polymorphs
http://www.google.com/patents/US20050209248
(S)-N-[[3-fluoro-4-[N-1[4-{2-furyl-(5-nitro)methyl}]piperazinyl]-phenyl]-2-oxo-5-oxazolidinyl]-methyl]acetamidehydrochloride having the Formula I.

The compound of Formula I, namely, (S)-N-[[3-fluoro-4-[N-1 [4-{2-furyl-(5-nitro)methyl}] piperazinyl]-phenyl]-2-oxo-5-oxazolidinyl]-methyl]acetamide hydrochloride is a phenyl oxazolidinone derivative, as disclosed in PCT application WO 02/06278. It is said to be useful as antimicrobial agent, effective against a number of human and veterinary pathogens, including gram-positive aerobic bacteria, such as multiply resistant staphylococci, streptococci and enterococci as well as anaerobic organisms such as Bacterioides spp. andClostridia spp. species, and acid fast organisms such as Mycobacterium tuberculosis, Mycobacterium avium and Mycobacterium spp.
The PCT application WO 02/06278 describes the preparation of compounds of Formula I. The products of Formula I obtained by following the cited methods tend to be hygroscopic and difficult to filter. These types of disadvantageous properties have proven to be serious obstacles to the large-scale manufacture of a compound. Further, handling problems are encountered during the preparation of pharmaceutical compositions comprising the hygroscopic compound of Formula I obtained by following the method disclosed in WO 02/06278.
EXAMPLE 1 Preparation of Polymorphic ‘Form A’ of the Compound of Formula I
50 gm of free base of Formula I was dissolved in ethanol (750 ml) by heating at about 60° C. and to this solution was added ethanolic HCl (13.36 ml, 8.9 N) at about 45-50° C. The reaction mixture was cooled to about 10° C., and stirred for about 4 hours. The separated solid was filtered off and dried under vacuum at 60° C. The solid was then digested in ethanol (150 ml) at 70-80° C. for about 4 hours. It was then cooled to about 10° C., the solid was filtered and dried under vacuum at 60-65° C. to give 30 gm of the pure polymorphic ‘Form A’ of compound of Formula I.
..................
Synthesis and SAR of novel oxazolidinones: Discovery of ranbezolid
Bioorg Med Chem Lett 2005, 15(19): 4261
http://www.sciencedirect.com/science/article/pii/S0960894X05008310
Synthesis and SAR of novel oxazolidinones: Discovery of ranbezolidPages 4261-4267Biswajit Das, Sonali Rudra, Ajay Yadav, Abhijit Ray, A.V.S. Raja Rao, A.S.S.V. Srinivas, Ajay Soni, Suman Saini, Shalini Shukla, Manisha Pandya, Pragya Bhateja, Sunita Malhotra, Tarun Mathur, S.K. Arora, Ashok Rattan, Anita Mehta | ||
Graphical abstract
Novel oxazolidinones were synthesized containing a number of substituted five-membered heterocycles attached to the ‘piperazinyl–phenyl–oxazolidinone’ core of eperezolid. Further, the piperazine ring of the core was replaced by other diamino-heterocycles. These modifications led to several compounds with potent activity against a spectrum of resistant and susceptible Gram-positive organisms, along with the identification of ranbezolid (RBx 7644) as a clinical candidate.Substitution of five-membered heterocycles on to the ‘piperazinyl–phenyl–oxazolidinone’ core structure led to the identification of ranbezolid as a clinical candidate. Further replacement of piperazine ring with other diamino-heterocycles led to compounds with potent antibacterial activity.
Scheme 5.
Reagents and conditions: (a) Method A: TFA, CH2Cl2, 0 °C → rt; 5-chloromethyl-2-furaldehyde, potassium carbonate, DMF, rt; or (b) Method B: TFA, CH2Cl2, 0 °C → rt; 5-nitrofuran-2-carboxaldehyde, sodiumtriacetoxyborohydride, THF, molecular sieves 3 Å, rt. 7 = ranbezolid
- Synthesis of compound 7: (S)-N-[[3-[3-Fluoro-4-(N-4-tert-butoxycarbonyl-piperazin-1-yl)phenyl]-2-oxo-5-oxa-zolidinyl]-methyl]acetamide (28a, 3.65 kg, 8.37 mol) was dissolved in dichloromethane (30.86 L) and cooled to 5 °C. To it trifluoroacetic acid (6.17 L) added dropwise and stirred for 14 h allowing the reaction mixture to warm to rt. The reaction mixture was evaporated in vacuo and the residue dissolved in tetrahydrofuran (58 L) followed by addition of molecular sieves 4 Å (4.2 kg). To the resulting mixture 5-nitro-2-furaldehyde (1.5 kg, 10.77 mol) was added followed by sodium triacetoxyborohydride (5.32 kg, 25.1 mol) and stirred for 14 h. The reaction mixture was filtered over Celite and filtrate evaporated in vacuo. The residue was dissolved in ethylacetate (85.6 L) and washed with satd sodium bicarbonate solution (36 L) and water (36 L). The organic layer was dried over anhyd sodium sulfate (3 kg) and evaporated in vacuo. The crude residue was purified by column chromatography (1–3% methanol in ethylacetate) to obtain (S)-N-[[3-[3-fluoro-4-[N-4-(5-nitro-2-furylmethyl)-piperazin-1-yl]phenyl]-2-oxo-5-oxa-zolidinyl]methyl]acetamide (39, 2.6 kg, yield 67%). Mp: 136 °C. 1H NMR (CDCl3): δ 7.42 (dd, 1H, phenyl–H), 7.29 (m, 2H, furyl–H), 7.07 (d, 1H, phenyl–H), 6.92 (t, 1H, phenyl–H), 6.51 (d, 1H, furyl–H), 6.11 (t, 1H, –NHCO–), 4.77 (m, 1H, oxazolidinone ring C5–H), 4.01 (t, 1H), 3.85–3.45 (m, 5H), 3.09 (m, 4H, piperazine–H), 2.72 (m, 4H, piperazine–H), 2.02 (s, 3H, –COCH3). MS m/z (rel. int.): 462.1 [(M+H)+, 100%], 484 [(M+Na)+, 25%], 500.2 [(M+K)+, 20%]. HPLC purity: 98%.
- Compound 39(3.6 kg, 7.81 mol) was dissolved in abs ethanol (53.8 L) by heating to 60 °C. The resulting solution was cooled to 45 °C and ethanolic hydrochloride (1.48 L, 7.9 N) was added dropwise in 10 min. The mixture was then cooled to 10 °C and stirred for 4 h and the precipitate formed was filtered and washed with ethanol and dried to obtain (S)-N-[[3-[3-fluoro-4-[N-4-(5-nitro-2-furylmethyl)-piperazin-1-yl]phenyl]-2-oxo-5-oxazolidinyl]-methyl]acetamide hydrochloride, ranbezolid (7, 3.2 kg, yield from 39: 82%, yield from 28a: 55%).
- Ranbezolid
- Mp: 207–209 °C.
- 1H NMR (DMSO, 300 MHz): δ 8.30 (t, 1H, –NHCO–), 7.75 (d, J = 3.3 Hz, 1H, furyl–H), 7.52 (dd, 1H, phenyl–H), 7.3–7.0 (m, 3H, phenyl–H, furyl–H), 4.70 (m, 1H, oxazolidinone ring C5–H), 4.63 (s, 2H), 4.08 (t, J = 8.8 Hz, 1H, –CH2–), 3.73 (t, J = 7.5 Hz, 1H), 3.43 (br m, piperazine–H merged with H2O in DMSO), 1.83 (s, 3H, –COCH3).
- HPLC purity: 98%. Anal. Calcd for C21H25ClN5O6·0.5H2O: C, 50.76; H, 5.48; N, 14.09. Anal. Found: C, 50.83; H, 5.17; N, 13.83.
References
- European Journal of Pharmacology. 2006. 545, 167–172
- US2005209248, 9-23-2005
Plymorphic forms of phenyl oxazolidinone derivatives - 1-1-2013Anti-anaerobic potential of ranbezolid: insight into its mechanism of action against Bacteroides fragilis.International journal of antimicrobial agents11-15-2009Synthesis and biological activity of novel oxazolidinones.Bioorganic & medicinal chemistry letters4-1-2009Mode of action of Ranbezolid against staphylococci and structural modeling studies of its interaction with ribosomes.Antimicrobial agents and chemotherapy8-1-2008Effect of oxazolidinone, RBx 7644 (ranbezolid), on inhibition of staphylococcal adherence to plastic surfaces.Journal of chemotherapy (Florence, Italy)4-1-2008Utilization of Bombyx mori larvae as a surrogate animal model for evaluation of the anti-infective potential of oxazolidinones.Journal of infection and chemotherapy : official journal of the Japan Society of Chemotherapy9-15-2007Synthesis and in vitro antibacterial activity of novel methylamino piperidinyl oxazolidinones.Bioorganic & medicinal chemistry letters9-18-2006Ranbezolid, a novel oxazolidinone antibacterial: in vivo characterisation of monoamine oxidase inhibitory potential in conscious rats.European journal of pharmacology10-1-2005Synthesis and SAR of novel oxazolidinones: discovery of ranbezolid.Bioorganic & medicinal chemistry letters6-1-2005Activity of RBx 7644 and RBx 8700, new investigational oxazolidinones, against Mycobacterium tuberculosis infected murine macrophages.International journal of antimicrobial agents10-1-2004In vitro activity of RBx 7644 (ranbezolid) on biofilm producing bacteria.International journal of antimicrobial agents
- 3-1-2003Antianaerobe activity of RBX 7644 (ranbezolid), a new oxazolidinone, compared with those of eight other agents.Antimicrobial agents and chemotherapy3-1-2003Antipneumococcal and antistaphylococcal activities of ranbezolid (RBX 7644), a new oxazolidinone, compared to those of other agents.Antimicrobial agents and chemotherapy
...................................
9 FYL 67

cas no 1416314-55-0
C20 H18 F N5 O3
FYL-67 IS HYDROCHLORIDE
(S)-N-((3-(3-Fluoro-4-(4-(pyridin-2-yl)-1H-pyrazol-1-yl)phenyl)-2-oxo-oxazolidin-5-yl)methyl)acetamide
N-[[(5S)-3-[3-fluoro-4-[4-(2-pyridinyl)-1H-pyrazol-1-yl]phenyl]-2-oxo-5-oxazolidinyl]methyl]-Acetamide,
(S)-N-((3-(3-fluoro-4-(4-(pyridin-2-yl)-1H-pyrazol-1-yl) phenyl)-2-oxooxazolidin-5-yl)methyl)acetamide.
| Inventores | Youfu LUO, 罗有福, Zhenling WANG, 王震玲,Yuquan Wei, 魏于全 |
| Requerente | Si Chuan University, 四川大学 |
The discovery and application of antibiotics is one of the greatest achievements of mankind in the 20th century, the field of medicine, called a revolution of the history of the human fight against illness. Since then, the field of medicine into a bacterial disease caused by greatly reducing the golden age. Today, however, due to the widespread use of antibiotics or even abuse, the growing problem of bacterial resistance, humans are gradually approaching the "post-antibiotic era, the efficacy of antibiotics is gradually reduced. Clinical have been found on many new drug-resistant strains of methicillin-resistant Staphylococcus aureus (MRSA), vancomycin-resistant enterococci (VRE), penicillin-resistant Streptococcus pneumoniae (PRSP) has seriously jeopardize the clinical treatment , the number of varieties of drugs less.
The compounds of the oxazolidinone linezolid was in the United States in 2000, mainly used in clinical acquired pneumonia, soft tissue infections, can also be used for the surgical treatment of infectious diseases, bones, lungs, cerebrospinal fluid permeability pharmacokinetic and tissue concentrations. Domestic and foreign the oxazolidinone drug development is a hot field
WO 2012171479
http://www.google.st/patents/WO2012171479A1?cl=en
The object compound (S N-{[3 - (3 - fluoro-4 - (4 - (2 - pyridyl) pyrazol-yl) phenyl) -2 - oxo-oxazol the embankment -5 - yl] methanone yl}
Weigh 150mg of the compound (26f), was dissolved with 10 ml of anhydrous THF was added under nitrogen protection, an ice water bath 154.1 mg t-BuOLi, ice-water bath after stirring for 5 minutes, 149.9 mg Compound 11, followed by ice-water bath was removed, go reaction at room temperature for 36 hours the reaction was stopped, by adding 10 mL of methylene chloride and 10 ml of water and 22μί acetic acid, stirred for 1 minute, the liquid separation, the aqueous phase was extracted with dichloromethane three times, the organic phases were combined, dried and purified by column chromatography to give the product ( 130 white solid 58 mg of yield of 38.2%.
1H-MR (400 MHz, CDC1 3): δ 8.61 (d, J = 4Hz, IH), 8.52 (d, J = 6.8Hz, 2.4H), 8.22 (s, IH), 7.94 (t, J = 8.8 Hz, IH), 7.77-7.69 (m, 2H), 7.55 (d, J = 8Hz, IH), 7.27-7.26 (m, IH), 7.18-7.15 (m, IH), 6.06 (t, J = 6Hz , IH), 4.86-4.80 (m, IH), 4.11 (t, J = 9.2Hz, IH), 3.86-3.82 (m, IH), 3.78-3.62 (m, 2H), 2.04 (s, 3H ;) .
13 C-MR (DMSO-e): δ 170.51, 154.47, 152.94, 151.26, 149.94, 139.70, 139.15, 137.43 129.96, 125.61, 125.19, 123.42, 122.19, 120.38, 114.52, 106.68, 72.29, 47.70, 41.84, 22.91.
ESI-MSm / z 418.08 (M + Na +).
......................
Nanoscale (2013), 5(1), 275-283
Carrier-free nanoassemblies of a novel oxazolidinone compound FYL-67 display antimicrobial activity on methicillin-resistant Staphylococcus aureus
Show Affiliations
a
State Key Laboratory of Biotherapy and Cancer Center, West China Hospital, West China Medical School, Sichuan University, Chengdu, China
E-mail: luo_youfu@scu.edu.cn, wangzhenling2007@126.com;
Fax: +86-28-85164060 ;
Tel: +86-28-85164063
E-mail: luo_youfu@scu.edu.cn, wangzhenling2007@126.com;
Fax: +86-28-85164060 ;
Tel: +86-28-85164063
b
Department of Gynecology and Obstetrics, Second West China Hospital, Sichuan University, Chengdu 610041, China
Nanoscale, 2013,5, 275-283
DOI: 10.1039/C2NR32505E
http://pubs.rsc.org/en/content/articlehtml/2013/nr/c2nr32505e
In this work, a novel oxazolidinone compound FYL-67 was synthesized, and the obtained FYL-67 could form nanoassemblies in aqueous solution by a self-assembly method without using any carrier, organic solvent, or surfactant. The prepared FYL-67 nanoassemblies had a particle size of 264.6 ± 4.3 nm. The FYL-67 nanoassemblies can be lyophilized into a powder form without any cryoprotector or excipient, and the re-dissolved FYL-67 nanoassemblies are stable and homogeneous. The in vitro release profile showed a significant difference between rapid release of free FYL-67 and much slower and sustained release of FYL-67 nanoassemblies. In vitro susceptibility tests were conducted in three strains of methicillin-susceptibleStaphylococcus aureus (MSSA) and three strains of methicillin-resistant Staphylococcus aureus(MRSA), using linezolid as a positive control. FYL-67 nanoassemblies exhibited excellent in vitro activity, with a minimum inhibitory concentration (MIC) value of 0.5 μg mL−1 against MRSA. In the in vitro post-antibiotic effect (PAE) evaluation, FYL-67 nanoassemblies showed a more powerful effect than linezolid. Besides, in vitro cytotoxicity tests indicated that FYL-67 nanoassemblies had a very low cytotoxicity on HEK293 cells and L02 cells. Furthermore, in both MSSA and MRSA systemic infection mouse models, FYL-67 nanoassemblies showed a lower ED50 than linezolid. In a murine model of MRSA systemic infection, FYL-67 nanoassemblies displayed an ED50 of less than 4.0 mg kg−1, which is 2.3-fold better than that oflinezolid. Our findings suggested that the FYL-67 nanoassemblies may be a potential drugcandidate in MRSA therapy.
 | ||
| Fig. 1 Synthetic route of the novel compound FYL-67. (i) 2-(pyridin-2-yl)malonaldehyde, p-TsOH (cat.),ethanol, reflux, 2 h; (ii) Fe, HCl, 95% ethanol, 1 h; (iii) Cbz–Cl, K2CO3, CH2Cl2, 2 h; (iv) (S)-1-acetamido-3-chloropropan-2-yl acetate, LiOt-Bu, THF, r.t.; (v) HCL (g), acetone, ethyl ether | ||
Synthesis of (S)-N-((3-(3-fluoro-4-(4-(pyridin-2-yl)-1H-pyrazol-1-yl) phenyl)-2-oxooxazolidin-5-yl)methyl)acetamide.
Benzyl(3-fluoro-4-(4-(pyridin-2-yl)-1H-pyrazol-1-yl)phenyl) carbamate (150 mg) was dissolved in absolute tetrahydrofuran under a nitrogen atmosphere in an ice bath. After stirring for 5 minutes, (S)-1-acetamido-3-chloropropan-2-yl acetate (149.9 mg) was added. The reactant was stirred at room temperature for another 36 hours. Then a mixture of dichloromethane (10 mL), distilled water (10 mL) and glacial acetic acid (0.022 mL) was added in order. The dichloromethane phase was collected using a separation funnel. The water phase was extracted with dichloromethane (10 mL) for another 2 times. The organic layer was combined and dried with anhydrous sodium sulfate. After removal of thesolvent, the residue was purified by flash chromatography and the title compound (58 mg) was obtained in a yield of 38.2%.
1H-NMR (400 MHz, CDCl3): δ 8.61 (d, J = 4 Hz, 1H), 8.52 (d, J = 6.8 Hz, 2.4H), 8.22 (s, 1H), 7.94 (t, J = 8.8 Hz, 1H), 7.77–7.69 (m, 2H), 7.55 (d, J = 8 Hz, 1H), 7.27–7.26 (m, 1H), 7.18–7.15 (m, 1H), 6.06 (t, J = 6 Hz, 1H), 4.86–4.80 (m, 1H), 4.11 (t, J = 9.2 Hz, 1H), 3.86–3.82 (m, 1H), 3.78–3.62 (m, 2H), 2.04 (s, 3H).
13C-NMR (DMSO-d6): δ 170.51, 154.47, 152.94, 151.26, 149.94, 139.70, 139.15, 137.43, 129.96, 125.61, 125.19, 123.42, 122.19, 120.38, 114.52, 106.68, 72.29, 47.70, 41.84, 22.91.
2.2.5. Prepration of FYL-67. 25 mg of (S)-N-((3-(3-fluoro-4-(4-(pyridin-2-yl)-1H-pyrazol-1-yl) phenyl)-2-oxooxazolidin-5-yl)methyl)acetamide was put in a 25 mL round-bottom flask, and 10 mL of acetonewas then added. After stirring for 5 minutes, the mixture turned transparent. Ethyl ether saturated with anhydrous hydrogen chloride was dropped in, and a white precipitate appeared. The collected yellowish powder was dried in a vacuum and 24.1 mg of powder was obtained with a yield of 88.3%.
1H-NMR (400 MHz, DMSO-d6) δ: 9.33 (s, 1H), 8.80 (s, 1H), 8.74 (d, J = 5.6 Hz, 1H), 8.45 (t, J = 7.2 Hz, 1H), 8.38–8.31 (m, 2H), 7.90 (t, J = 8.8 Hz, 1H), 7.81 (dd, J = 2.4 Hz, J = 16.4 Hz, 1H), 7.76 (t,J = 6.0 Hz, 1H); 7.55 (dd, J = 1.6 Hz, J = 8.8 Hz, 1H), 4.83–4.76 (m, 1H), 4.60 (br s, 1H), 4.20 (t, J = 8.8 Hz, 1H), 3.91–3.82 (m, 1H), 3.45 (t, J = 5.2 Hz, 2H), 1.85 (s, 3H);
13C-NMR (DMSO-d6) δ: 170.51, 154.47, 152.94, 151.26, 149.94, 139.70, 139.15, 137.43, 129.96, 125.61, 125.19, 123.42, 122.19, 120.38, 114.52, 106.68, 72.29, 47.70, 41.84, 22.91;
HR-MS(TOF) m/z calcd for C20H18FN5O3 [M + Cl−]: 430.1082, found: 430.1085; for C20H18FN5O3 [M + H+]: 396.1472, found: 396.1472.
.................................
PAPER
Org. Process Res. Dev., 2014, 18 (4), pp 511–519
DOI: 10.1021/op500030v
A concise, environmentally benign, and cost-effective route was developed for the large-scale preparation of 1, a novel oxazolidinone antibacterial candidate. The key intermediate 2-(1-(2-fluoro-4-nitrophenyl)-1H-pyrazol-4-yl)pyridine 7 was prepared with high purity by mild deamination of the regioisomeric mixture 21. The mixture was prepared from a nucleophilic SNAr reaction by selective C–N coupling of the secondary amine functionality of 4-(pyridin-2-yl)-1H-pyrazol-3-amine 14 with 1,2-difluoro-4-nitrobenzene 10 in optimized conditions with the primary amine group remaining intact. The gaseous nitrogen release rate and reaction mixture temperature of the deamination step can be well controlled by altering the feeding manner, thereby providing safety guarantees. The optimized synthetic strategy of 1 with an overall yield of 27.6%, including seven sequential transformations by only five solid–liquid isolations, significantly improved the product separation workup. The strategy bypassed time-consuming and laborious procedures for any intermediate involved as well as for the final API. This study presents a process enabling the rapid delivery of a multikilogram quantity of API with high purity.
\
(S)-N-((3-(3-Fluoro-4-(4-(pyridin-2-yl)-1H-pyrazol-1-yl)phenyl)-2-oxo-oxazolidin-5-yl)methyl)acetamide (1)
In a 50-L reactor, 9 (1.8 kg, 4.64 mol) and 8 (1.79 kg, 9.27 mol) were dissolved in THF (12.6 L) at −5 °C. The reaction mixture was degassed by purging with N2. Then, methanol (375 mL, 9.27 mol) was added to the mixture under N2 atmosphere. After stirring for about 10 min at −5 °C, lithium tert-amylate (1.11 kg, 13.91 mol) was added to the mixture in one portion with an exotherm from −5 to 17 °C. The resulting solution was cooled to −5 °C, yielding a thick slurry, and stirred for about 1 h and stirred again at 25 °C for about 15 h. The slurry was cooled to 10 °C. The reaction was quenched by adding acetic acid (525 mL, 9.27 mol) in one portion and stirred for 30 min. The reaction mixture was then evaporated to dryness at 30 °C. The solid residue was allowed to soak for 3 h in water (30 L), stirred, filtered under reduced pressure, and washed with water (10 L × 3). The solid filtered cake was suspended in ethyl acetate (10 L). The resulting suspension was heated to reflux for 2 h, cooled to 25 °C, and filtered under reduced pressure. The collected solid was resuspended in a mixture of EtOH and water (6 L/2 L) and heated to reflux for 2 h. The slurry was cooled to 25 °C, filtered under reduced pressure, and washed with EtOH (3 L × 2). The filtered cake was dried in an oven to a constant weight at 45 °C. The final product was an off-white solid 1 (1.5 kg, isolated yield of 82%).
The HPLC purity was over 99.9%.
1H NMR (400 MHz, CDCl3): δ 8.61 (d, J = 4 Hz, 1 H), 8.52 (d, J = 6.8 Hz, 2 H), 8.22 (s, 1 H), 7.94 (t, J = 8.8 Hz, 1 H), 7.77–7.69 (m, 2 H), 7.55 (d, J = 8 Hz, 1 H), 7.27–7.26 (m, 1 H), 7.18–7.15 (m, 1 H), 6.06 (t, J = 6 Hz, 1 H), 4.86–4.80 (m, 1 H), 4.11 (t, J = 9.2 Hz, 1 H), 3.86–3.82 (m, 1 H), 3.78–3.62 (m, 2 H), 2.04 (s, 3 H);
13C NMR (DMSO-d6): δ 170.51, 154.47, 152.94, 151.26, 149.94, 139.70, 139.15, 137.43, 129.96, 125.61, 125.19, 123.42, 122.19, 120.38, 114.52, 106.68, 72.29, 47.70, 41.84, 22.91;
ESI-MS m/z 418.08 (M + Na+).
- Brickner, S. J.; Hutchinson, D. K.; Barbachyn, M. R.; Manninen, P. R.; Ulanowicz, D. A.; Garmon, S. A.; Grega, K. C.; Hendges, S. K.; Toops, D. S.; Ford, C. W.; Zurenko, G. E.J. Med. Chem. 1996, 39, 673– 679
(b) Barbachyn, M. R.; Ford, C. W. Angew. Chem., Int. Ed. 2003, 42, 2010– 2023
- (a) Gong, C. Y.; Yang, T.; Yang, X. Y.; Liu, Y. Y.; Ang, W.; Tang, J. Y.; Pi, W. Y.; Xiong, L.; Chang, Y.; Ye, W. W.; Wang, Z. L.; Luo, Y. F.; Zhao, X.; Wei, Y. Q. Nanoscale. 2013, 5, 275–283
(b) Luo, Y. F.; Wang, Z. L.; Wei, Y. Q.; Geng, F. WO/2012/171479,2012.WO2008143649A2 * 4 Dez 2007 27 Nov 2008 Das Jagattaran Novel oxazolidinone compounds as antiinfective agents CN1172484A * 29 Jan 1996 4 Fev 1998 法玛西雅厄普约翰美国公司 Hetero-aromatic ring substituted phenyloxazolidinone antimicrobials
10
WCK ?........http://newdrugapprovals.org/2015/11/24/new-antibacterial-oxazolidinones-in-pipeline-by-wockhardt/
WCK ?
(5S)-N-{3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide
(5S)-N- {3-[3,5-difluoro-4-(4-hydroxy-(4-methoxymethyl)-piperidin- lyl)phenyl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide
MF C19 H25 F2 N3 O5, MW 413.42
Acetamide, N-[[(5S)-3-[3,5-difluoro-4-[4-hydroxy-4-(methoxymethyl)-1-piperidinyl]phenyl]-2-oxo-5-oxazolidinyl]methyl]-
CAS 957796-51-9
Antibacterial oxazolidinones
Wockhardt Ltd, Innovator
Wockhardt Research Center,
THIS MAY BE WCK 4086?????….WATCHOUT THIS POST FOR UPDATION
PATENTSWO 2015173664, US8217058, WO 2012059823, IN 2011MU03726

Oxazolidinone represent a novel chemical class of synthetic antimicrobial agents. Linezolid represents the first member of this class to be used clinically. Oxazolidinones display activity against important Gram-positive human and veterinary pathogens including Methicillin-Resistant Staphylococcus aureus (MRSA), Vancomycin Resistant Enterococci (VRE) and β-lactam Resistant Streptococcus pneumoniae (PRSP). The oxazolidinones also show activity against Gram-negative aerobic bacteria, Gram-positive and Gram-negative anaerobes. (Diekema D J et al., Lancet 2001 ; 358: 1975-82).
Various oxazolidinones and their methods of preparation are disclosed in the literature. International Publication No. WO 1995/25106 discloses substituted piperidino phenyloxazolidinones and International Publication No. WO 1996/13502 discloses phenyloxazolidinones having a multisubstituted azetidinyl or pyrrolidinyl moiety. US Patent Publication No. 2004/0063954, International Publication Nos. WO 2004/007489 and WO 2004/007488 disclose piperidinyl phenyl oxazolidinones for antimicrobial use.
Pyrrolidinyl/piperidinyl phenyl oxazohdinone antibacterial agents are also described in Kim H Y et al., Bioorg. & Med. Chem. Lett., (2003), 13:2227-2230. International Publication No. WO 1996/35691 discloses spirocyclic and bicyclic diazinyl and carbazinyl oxazolidinone derivatives. Diazepeno phenyloxazolidinone derivatives are disclosed in the International Publication No. WO 1999/24428. International Publication No. WO 2002/06278 discloses substituted aminopiperidino phenyloxazolidinone derivatives.
Various other methods of preparation of oxazolidinones are reported in US Patent No. 7087784, US Patent No. 6740754, US Patent No. 4948801 , US Patent No. 3654298, US Patent No. 5837870, Canadian Patent No. 681830, J. Med. Chem., 32, 1673 (1989), Tetrahedron, 45, 1323 (1989), J. Med. Chem., 33, 2569 (1990), Tetrahedron Letters, 37, 7937-40 (1996) and Organic Process Research and Development, 11 , 739-741(2007).
Indian Patent Application No. 2534/MUM/2007 discloses a process for the preparation of substituted piperidino phenyloxazolidinones. International Publication No. WO2012/059823 further discloses the process for the preparation of phosphoric acid mono-(L-{4-[(5)-5-(acetylaminomethyl)-2-oxo-oxazolidin-3-yl]-2,6-difluorophenyl}4-methoxymethyl piperidine-4-yl)ester.
US Patent No. 8217058 discloses (5S)-N-{3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-l-yl)-phenyl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide as an antibacterial agent and its process for preparation.
PATENT
WO2015173664https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015173664&recNum=1&maxRec=&office=&prevFilter=&sortOption=&queryString=&tab=PCTDescription
In some embodiments, there is provided a process for preparation of a compound of Formula (I) as shown in Scheme 1
(I I) (I N)
Scheme 1
Example 1
Preparation of (55)-iV-{3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-l-yl)- phenyl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide (I)
To a stirred solution of lithium teri-butoxide (59.1 g, 0.74 mol) in tetrahydrofuran (500 ml) was added a solution of [3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-l-yl)-phenyl]-carbamic acid benzyl ester (II) (100 g, 0.25 mol) in 500 ml of tetrahydrofuran slowly at room temperature. The resulting mixture was stirred for 3 hours at room temperature (formation of lumps observed). The reaction mixture was cooled to temperature of 10°C to 15°C and acetic acid l-(acetylamino-methyl)-2-chloro-ethyl ester (III) (95.2 g, 0.49 mol) was added in one lot, after 5 minutes methanol (2.36 g, 0.075 mol) was added in one portion. The resulting mixture was stirred further at temperature of 10°C to 15°C. After 5 hours the reaction mixture was allowed to warm to room temperature and stirring continued further for 16 hours. An aqueous solution of saturated ammonium chloride (100 ml) was added to the reaction mixture, the resulting mixture was stirred well and the solvent evaporated under reduced pressure (35°C, 150 mm Hg). The residual mixture was diluted with water (1 L stirred well and filtered under suction, the residual solid was washed with additional fresh water (100 ml). The residual mass was suspended in acetone (500 ml), stirred well and the mixture diluted with hexane (1 L), slowly. The mixture was stirred further for 1 hour and filtered under suction. The residual solid was washed with a 2:1 mixture of acetone and water (100 ml). The residual solid was dried at 45°C, for 3.5 hour at 4 mm Hg, to obtain the 78 g of (55)-N-{3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-l -yl)-phenyl]-2-oxo-oxazolidin-5-ylmethylj -acetamide (I) as white solid, in 77% yield.
Analysis:
Mass: 414 (M+l ); for Molecular Weight: 413 and Molecular Formula:
Melting Point: 178-179°C;
1H NMR (400 MHz, DMSO): δ 8.18-8.21 (m, 1H), 7.19-7.25 (d, 2H), 4.07-4.71 (m, 1H), 4.32 (s, 1H), 4.02-4.07 (t, 1H), 3.64-3.68 (t, 1H), 3.14 (s, 2H), 2.81-2.83 (d, 2H), 1.81 (s, 3H), 1.63-1.69 (t, 2H), 1.42-1.45 (d, 2H);
Purity as determined by HPLC: 97.65%.
Example 2
Preparation of acetic acid l-(acetylamino-methyl)-2-chloro-ethyl ester (III)
Step-I: Preparation of l-amino-3-chloro-propan-2-ol hydrochloride (VI)
Benzaldehyde (118.67 g, 1.03 mol) was dissolved in ethanol (297 ml) under stirring and the solution was cooled to 18-19°C. To this solution aqueous ammonia solution (25%) (101.58 ml) was added slowly, followed by slow addition of S-epichlorohydrin (100 g, 1 mol). The resulting mixture was warmed to 40°C and stirred for 7 hours. The mixture was allowed to cool to room temperature and stirred further. After 16 hours, the reaction mixture was concentrated to 50% volume under reduced pressure. Toluene (228 ml) was added to the reaction mixture followed by addition of aqueous hydrochloric acid (162 ml of concentrated hydrochloric acid diluted with 152 ml of water). The mixture thus obtained for 3 hours at 45°C, the resulting mixture was allowed to cool to room temperature and the toluene layer separated. The toluene layer was further extracted with water (56 ml). The combined aqueous layer was diluted with ethanol (56 ml) and the mixture evaporated under reduced pressure. This process was repeated again. To the final concentrate was added ethanol (180 ml), stirred for 10 minutes and the mixture cooled to -28°C to -30°C and maintained at this temperature for 2 hours. The separated solid was filtered under suction and the residue washed with cold (-30°C) ethanol (50 ml). The residue was dried at 45°C, under reduced pressure (4 mm Hg) for 3 hours, to obtain 96 g of l-amino-3-chloro-propan-2-ol hydrochloride (VI) as white solid in 61% yield.
Analysis:
Mass: 110 (M+l) as free base; for Molecular Weight: 145.5 and Molecular Formula:
1H NMR (400 MHz, D20): δ 4.02-4.08 (m, 1H), 3.51-3.61 (m, 2H), 3.12-3.16 (dd, 1H), 2.93 -2.99 (dd, 1H).
Step-II: Preparation of acetic acid l-(acetylamino-methyl)-2-chloro-ethyl ester (III).
A stirred solution of dichloromethane (220.8 ml) containing the step-I salt (96 g, 0.66 mol) was cooled to 18-20°C. Acetic anhydride (154.78 g, 1.5175 mol) was added slowly (slight exothermic). Pyridine (67.76 g, 0.8577 mol) was added slowly (exothermic) while maintaining the temperature at 18-20°C. The resulting mixture was heated to 40°C for 5 hours. The reaction mixture was allowed to cool to room temperature and stirring continued for further 16 hours. The reaction mass was cooled to 3-6°C and diluted with 170 ml of fresh water. To this was added an aqueous solution of potassium carbonate (191.2 g of K2CO3 in 382 ml water). The reaction mixture was further diluted with additional dichloromethane (170 ml) and water (425 ml). The reaction mass was stirred well and the dichloromethane layer separated. The aqueous layer was further extracted with 2×170 ml dichloromethane. The combined dichloromethane layer was washed with aqueous sodium chloride solution (13.6 g of sodium chloride in 493 ml water). The solvent was evaporated till a volume of 170 ml and the residual layer was diluted with toluene (340 ml), stirred well and the solvent was evaporated completely at 40°C under reduced pressure (4 mm Hg). To the residue ethyl acetate (170 ml) and hexane (187 ml) were added and the mixture stirred for 30 minute. The separated solid was filtered under suction and the residue washed with 50 ml of a 1 :1 mixture of ethyl acetate and hexane. The solid obtained was dried under reduced pressure (4 mm Hg) at 45°C for 3.5 hours, to obtain 96 g of acetic acid l-(acetylamino-methyl)-2-chloro-ethyl ester (III) as a white solid, in 75% yield.
Analysis:
Mass: 194 (M+l); for Molecular Weight: 193 and Molecular Formula: C7Hi2ClN03; 1H NMR (400 MHz, CDC13): 5 5.69 (s, 1H), 5.0-5.1 (m, 1H), 3.4-3.7 (m, 4H), 2.1 (s, 3H), 1.9 (s, 3H).
PATENT
http://www.google.st/patents/WO2007132314A2?cl=en
Wockhardt Ltd,


Scheme -1



Example -11 : (5S)-N- {3-[3,5-difluoro-4-(4-hydroxy-(4-methoxymethyl)-piperidin- lyl)phenyl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide
The example- 10 (54.86 g, 0.144 mol) was suspended in methanol (1100 ml) under stirring at RT. Sodium metal (4 g, 0.174 mol) was added in small lots in 2 min to the above suspension under stirring. The reaction mixture was warmed to 40-420C and was stirred at this temperature for about 40 hrs. After completion of the reaction (TLC), the solvent was evaporated under reduced pressure to obtain a thick slurry. The thick slurry thus obtained was gradually added to water (1100 ml) under stirring. After the complete addition, the pH of the aqueous suspension was adjusted to 7 by adding sufficient quantity of glacial acetic acid. The separated solid was filtered and the residue was washed with water. The obtained solid was further purified by column chromatography over silica gel to obtain the product as a white solid, 32.7 g, 55 % yield.
M.P.: 173-1740C;
MS : M+l= 414(MH+, 100%); for M.F.: Ci9H25F2N3O5
1H-NMR (400 MHz, CDCl3): δ 7.0-7.1 (m, 2H5Ar-H), 6.0 (t, IH, NH), 4.70-4.80 (m, IH), 4.00 (t,lH), 3.70-3.75 (m, 2H), 3.5-3.7 (m, IH), 3.43 (s, 3H, OCH3), 3.37-3.42 (m, 2H), 3.30 (s, 2H, -OCH2), 3.0-3.05 (m, 2H), 2.22(bs,lH ,-OH),2.04 (s, 3H, COCH3), 1.70-1.75 (m, 4H).
Patent
INDIAN 3049/MUM/2010Phosphoric acid mono-(1-{4-[(S)-5-(acetylamino-methyl)-2-oxo-oxazolidin-3-yl]-2,6-difluorophenyl}-4-methoxy methyl-piperidin-4-yl) ester

Specific intermediate compounds of the invention include:
6-(2,6-difluoro-4-nitrophenyl)-1-oxa-6-azaspiro[2.5]octane;
1-(2,6-Difluoro-4-nitro-phenyl)-4-methoxymethyl-piperidin-4-ol;
[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-carbamic acid benzyl ester;
(5R)-3-[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-5-hydroxymethyl-oxazolidin-2-one;
(5R)-Methanesulfonic acid 3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-2-oxo-oxazolidin-5-ylmethyl ester;
(5R)-3-[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-5-azidomethyl-oxazolidin-2-one; and
(5S)- N-{3-[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide.
Examples
Preparation of Intermediate-1: 1-(2,6-Difluoro-4-nitrophenyl)-piperidin-4-one
Chloroform (9.3 L) was charged in a 20 L reaction assembly and 4-piperidone hydrochloride (1.17 Kg, 7.62 mol) was added under stirring followed by triethylamine (2.14 Kg, 2.95 L, 21.1 mol). After 30 minutes of stirring, 3,4,5-trifluoronitrobenzene (1.5 Kg, 8.47 mol) was added to the mixture in one lot and the contents were heated to 65-70ºC for 8 h. After completion of the reaction, chloroform was removed under vacuum to obtain a syrupy mass. At this stage, water (10 L) was added to the mass and the chloroform recovery was continued under vacuum below 65oC till the chloroform was removed completely. The slurry was cooled to RT and filtered. The solid product was washed with water (3 L) followed by hexanes (2 L). The product was dried in a vacuum oven below 70oC to obtain the product as a yellow solid, 1.88 Kg ; Yield 97%.
M.P.: 130-132oC; MS: 257(M+1); M.F.: C11H10F2N2O3.
Preparation of Intermediate 3: 1-(2,6-Difluoro-4-nitro-phenyl)-4-methoxymethyl-piperidin-4-ol
Method A:
Preparation of Intermediate–2: (Stage-I): 6-(2,6-difluoro-4-nitrophenyl)-1-oxa-6-azaspiro[2.5]octane
A solution of trimethylsulfoxonium iodide (1.504kg, 6.836mol) in acetonitrile (7L) was cooled to 0 to 5oC. , under argon atmosphere. Potassium tert-butoxide (0.736kg, 6.552 mol) was added in small lots over 0.5h. The resulting solution was stirred for 2h at the same temperature. To this solution was added 1-(2,6-Difluoro-4-nitrophenyl)-piperidin-4-one ( 1.4kg, 5.46mol) in small lots over a period of 1h, while maintaining the temp. between 5-10oC. The resulting mixture was stirred for 1h. The solvent was evaporated to a minimum amount possible, under reduced pressure while maintaining the temperature below 10oC. The residue was poured in water( 18L) and the pH adjusted to neutral with dilute acetic acid. The resulting slurry was stirred well and the separated solid filtered under suction. The solid was washed with fresh water till the filtrate was free of acetic acid. The solid was dried at 80oC, for 6h, under reduced pressure to obtain the product as pale yellow solid, 1.264kgs, yield 85%.
M.P.: 96-97oC; MS: M+1: 271; M.F.: C12H12F2N2O3,.
Preparation of Intermediate-3: (Stage-II): 1-(2,6-Difluoro-4-nitro-phenyl)-4-methoxymethyl-piperidin-4-ol
To a solution of sodium methoxide (236g, 4.35mol) in methanol (3L), at RT, was added 6-(2,6-difluoro-4-nitrophenyl)-1-oxa-6-azaspiro [2.5]octane (964g, 3.57mol) in small portions and the reaction mixture was stirred for 26h at RT. Acetic acid (265g, 4.44mol) was added slowly to neutralize the pH of the solution. The resulting mixture was poured into chilled water(18L) and stirred for 1h. The separated solid was filtered under suction. The solid was washed with additional water till the filtrate was free of acetic acid. The solid was dried for 10hat RT under reduced pressure, to obtain the product as a pale yellow solid, 973g, yield, 90%
M.P.: 84-86oC; MS: 303 (M+1); M.F.: C13H16F2N2O4
Method B:
Dimethylsulfoxide (DMSO, 100 ml) and methanol (500 ml) were charged in a 1 L glass reaction assembly. Potassium hydroxide (59.2g, 0.898 mol) was charged in the assembly followed by trimethylsulfoxonium iodide (94.5 g, 0.43 mol) and the contents were stirred for 30 minutes and then cooled to 10oC-15oC. To the cooled contents was added 1-(2,6-difluoro-4-nitrophenyl)-piperidin-4-one (100 g, 0.39 mol) in small lots. After the addition, the temperature was allowed to raise to RT and the contents were further stirred for 24 h (ring opening of the epoxide intermediate viz. 6-(2,6-difluoro-4-nitrophenyl)-1-oxa-6-azaspiro[2.5]octane takes place).
[Physical data of the intermediate: M.P.: 96-970C, MS: 271(M+1); M.F.: C12H12F2N2O3, .
After completion of the reaction the contents were poured slowly in ice-water (600g crushed ice in 600 ml water). The precipitated solid product was filtered and was washed with water:methanol, 2:1 (100 ml X 2). The wet product was used in the next step.
M.P.: 84-86oC; MS: 303 (M+1);.M.F.: C13H16F2N2O4,:
Preparation of Intermediate -5: [3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-carbamic acid benzyl ester
Method A: Preparation of Intermediate 4: ( Stage-I)
Water (1.19 L) and methanol (595 ml) were charged in a 3 L glass reaction assembly, followed by 1-(2,6-difluoro-4-nitro-phenyl)-4-methoxymethyl-piperidin-4-ol (85 g, 0.281 mol) and the contents were stirred. Sodium dithionite (288 g, 1.407 mol) was added in one lot and the reaction mixture was heated to 80oC for 8 h. After completion of the reaction (TLC), methanol was recovered under vacuum below 65oC. After the recovery, the aqueous residue was extracted with chloroform (400 ml X 3). The combined chloroform extract (containing the intermediate 1-(4-amino-2,6-difluoro-phenyl)-4-methoxymethyl-piperidin-4-ol) was dried over anhydrous Sodium sulfate and used in the next step (carbamate formation).
Preparation of Intermediate -5: (Stage-II):
The above chloroform extract was charged in a 3 L glass reaction assembly. Sodium bicarbonate (70 g, 0.843 mol) was added to the extract and the contents were cooled to 15oC-20oC. Benzylchloroformate solution (50% in toluene, 48 g, 96 ml, 0.281 mol) was added slowly to the above mixture under stirring. After completion of the addition, the reaction mixture was stirred at RT for 2 h. After completion of the reaction (TLC), the contents were filtered on a Buchner assembly and the solid cake was washed with chloroform (85 ml X 2). The combined filtrate was evaporated under vacuum below 50oC to obtain yellowish oily mass, which was poured slowly in hexanes (850 ml) under stirring to obtain a precipitate. The precipitated product was filtered and washed with hexanes (100 ml X 2). The product was dried in a vacuum oven below 65oC to obtain 60.2 g brownish product (Yield = 38% on the basis of step-I input).
M.P.: 138-140oC; MS: 407(M+1); M.F.: C21H24F2N2O4.:.
Method B: : Preparation of Intermediate 4: ( Stage-I): To a solution of 1-(2,6-difluoro-4-nitro-phenyl)-4-methoxymethyl-piperidin-4-ol (973g, 3.22 mol) in ethyl acetae (10L) was added 10% Pd-C, (250g, 50% wet) and the resulting miture was hydrogenated in a pressure at 30 PSI, 45-55oC, for 3h. The catakyst was filtered and the residue was washed with additional ethyl acetate( 200ml). The combined filtrates were used as such for the next reaction (carbamate formation)
Preparation of Intermediate -5: (Stage-II):
To the above filtrate was added sodium bicarbonate(406g, 4.83 mol) and the mixture warmed to 40-45oC. To this mixture was added a 50% solution of Benzyl chloroformate in toluene(1.373L, 4.025 mol), drop-wise, over a period of 1h. Stir the resulting mixture for 1h and filter the insoluble material. The residue was washed with 300ml of ethyl acetate. The filtrates were combined and the solvent evaporated under reduced pressure, below 55oC.. Cool the residue and dilute it with hexane(10L). The resulting slurry was stirred well and the separated solid was filtered under suction. The residue was washed with additional hexane ( 2L). The solid was dried for 10h at RT, to obtain the product as dark brown solid, 1200g, yield, 96%.
M.P.: 138-140oC; MS: 407( M+1); M.F.: C21H24F2N2O.
Preparation of Intermediate -6:
(5R)-3-[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-5-hydroxymethyl-oxazolidin-2-one
To a mixture of [3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-carbamic acid benzyl ester (100g, 0.237 mol) in dry tetrahydrofuran (THF) (2 L) at 40ºC was added drop-wise n-BuLi in hexane (1.6M, 45.5 g, 455 ml, 0.711 mol) under nitrogen atmosphere. The contents were stirred for 1 h at 40ºC and R-(-)-glycidyl butyrate (68.25 g, 0.474 mol) was added gradually. After the addition of R-(-)-glycidyl butyrate, the reaction mixture was stirred for 5-6 h at 40oC till completion of the reaction (TLC). After completion of the reaction, a solution of sodium methoxide (2 g) in methanol (66 ml) was added to the contents followed by water (8 ml) and the contents were stirred for an additional 0.5 h. Water (1 L) was added to the solution and the contents were extracted with ethyl acetate (1 L). The aqueous layer was further extracted with ethyl acetate (3 X 500 ml). The combined organic layer was evaporated under vacuum to obtain a thick residue. tert-Butyl methyl ether (1 L) was added to the residue and the contents were stirred for about 1 h to obtain a solid product, which was filtered and washed with tert-butyl methyl ether (2 X 100 ml). The product was dried under vacuum below 60ºC to obtain the product as a 46.5 g dark brown compound, 46.5g ,yield 51%.
M.P.: 117-119oC; MS: 373(M+1); M.F.: C17H22F2N2O5..
Preparation of Intermediate -7: (5R)-Methanesulfonic acid 3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-2-oxo-oxazolidin-5-ylmethyl ester
To a mixture of (5R)-3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-5-hydroxymethyl-oxazolidin-2-one (45 g, 0.121 mol) in dichloromethane (0.3 L), was added triethylamine (24.5 g, 34 ml, 0.242 mol) while stirring. Methanesulfonyl chloride (18 g, 12.2 ml, 0.157 mol) was added to the above solution over a period of 1 h at 10oC -20oC and the reaction mixture was stirred for additional 2 h at RT. After completion of the reaction (TLC), the contents were evaporated under vacuum at 40oC to obtain an oily residue. Water (450 ml) was added to the residue and the traces of dichloromethane were removed under vacuum. The solid product thus obtained was filtered, washed with water (2 X 50 ml) and dried under vacuum at 70oC to obtain 50.6 g brownish compound. Yield = 93%; M.P.:106-108oC; MS: 451(M+1); M.F.: C18H24F2N2O7S.
Preparation of Intermediate 8a: (5R)-3-[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-5-azidomethyl-oxazolidin-2-one
Method A:
To a solution of (R)-3-(3,5-difluoro-4-(4-hydroxy-4-(methoxymethyl)piperidin-1-yl)phenyl)-5-(hydroxymethyl)oxazolidin-2-one (2g, 5.3 mmol),in tetrahydrofuran (20 mL), under argon , was added diphenylphosphoryl azide (1.63mL, 5.9 mmol). The solution was cooled to 0oC in an ice-bath. 1,8-diazabicyclo [5.4.0] undec-7-ene (DBU) (0.76mL, 4.9mmol) was added drop-wise over 15min..The reaction was stirred at same temperature for 1 hr, and then warmed to room temperature and stirred under for 16 hr. The reaction mixture was diluted with ethyl acetate (20 mL), and water (20mL). After separation of water layer, the organic layer was washed with water and 0.5M citric acid monohydrate (10 mL). The organic layer was dried over sodium sulfate and the solvent evaporated under reduced pressure.The residue was triturated with ether to obtain the product as a buff colored solid, 1.32g (62%).
M.P.: 106-108oC; M.S.- 398(M+1); M.F.- C17H21F2N5O4,
Method B:
To a solution of (5R)-methanesulfonic acid 3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-2-oxo-oxazolidin-5-ylmethyl ester (20 g, 0.044 mol, wet) in N,N-dimethylformamide (30 ml), was added sodium azide (8.6 g, 0.133 mol) in a single lot. The reaction mixture was gradually heated and the temperature was maintained at 70ºC for 8 h. After completion of the reaction (TLC), the contents were cooled to 20-25ºC and poured slowly into chilled water (300 ml). The solid product thus obtained was filtered and washed with water (2 x 50 ml). The wet product was air dried to obtain 16.5g dark brown compound (being an azide, it was NOT exposed to heat during drying) Yield ~ 93%.
M.P.: 106-108oC; MS : 398(M+1); M.F.: C17H21F2N5O4;:
Preparation of Intermediate 8b: (5S)-N-2-{3-[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-2-oxo-oxazolidin-5-ylmethyl}-phthalimide
Method A:
A mixture of (5R)-{3-[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)phenyl]-2-oxo-oxazolidin-5-yl methyl}-methanesulfonate(10g, 0.022 mol), Potassium phthalimide (12.2g, 0.066 mol) and DMF (50ml) was heated, with stirring, at 90oC for 4h. The resulting mixture was cooled to RT and poured over ice-water mixture. The separated solid was filtered, washed with water and dried under suction to obtain the product as a white solid, 9.46g, in 85% yield.
M.P.: 154-156 oC; MS: 502 (M+1); M.F. C25H25F2N3O6.
Method B:
To tetrahydrofuran (30 ml) were added triphenylphosphine (2.11g, 8 mmol)) and diethyldiazocarboxylate (1.62g, 8 mmol)), and the solution stirred at room temperature. After 10 minute phthalimide (1.18g, 8 mmol)) was added and after a further stirring for 10 minute, (R)-3-(3,5-difluoro-4-(4-hydroxy-4-(methoxymethyl)piperidin-1-yl)phenyl)-5-(hydroxymethyl) oxazolidin-2-one (2g, 5.3 mmol) was added and stirring continued further at room temperature. After 8 hrs ice-cold water (4 ml) was added to the reaction mixture and the resulting mixture was extracted by ethyl acetate (2 x 20ml). The ethyl acetate extract was dried (over sodium sulfate) and concentrated under reduced pressure. The residue was chromatographed on a column of silica gel to obtain the product as an off-white solid, 1.56g, yield 58%.
M.P.: 154-156 oC; MS : 502 (M+1); M.F. C25H25F2N3O6.
Preparation of Intermediate 10: (5S)- N-{3-[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide
via
Intermediate 9: 5-aminomethyl-3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-oxazolidin-2-one
Method A:
To a solution of (5R)-3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-5-azidomethyl-oxazolidin-2-one (10 g, 0.025 mol) in methanol (100 ml), were charged cobalt chloride (0.6 g, 0.0025 mol) followed by sodium borohydride (0.95 g, 0.025 mol) in small lots over a period of 30 minutes. The reaction mixture was stirred at RT for additional 2 h. After completion of the reaction , the contents were evaporated under vacuum below 40oC to obtain a sticky mass. The contents were suspended in a mixture of water (100 ml) and ethyl acetate (50 ml) and stirred for 15 minutes. The contents were filtered through a filter-aid bed and the bed was washed with ethyl acetate (2 X 25 ml). The layers were separated and the aqueous layer was further extracted with ethyl acetate (4 X 50 ml). The combined organic layer was washed with 1% HCl solution (100 ml). The aqueous layer was separated and washed with dichloromethane (4 X 50 ml). The pH of the aqueous layer was adjusted to 8 by adding saturated sodium bicarbonate solution. The contents were extracted with ethyl acetate (6 X 50 ml) till no amine spot was seen in the final organic extract. The combined organic layer (containing the intermediate 5-aminomethyl-3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-oxazolidin-2-one) was dried over anhydrous sodium sulfate.
Triethylamine (3.3 g, 4.5 ml, 0.0327 mol) was added to the above organic layer and acetyl chloride (2.17 g, 2 ml, 0.0277 mol) was added gradually over a period of 1 h at RT. The reaction mixture was stirred for 2 h and after completion of the reaction (TLC), the contents were washed with water (50 ml) and the layers separated. Activated carbon (1 g) was added to the organic layer and the contents were stirred for 15 minutes. The contents were filtered on a celite bed and the carbon-celite bed was washed with ethyl acetate (2 X 10 ml). The combined filtrate was evaporated under vacuum to obtain a slurry, which was filtered on a Buchner assembly and the product was washed with ethyl acetate (2 X 10 ml). The product was dried under vacuum at 70oC to obtain 5 g off-white solid. Yield = 48% (on the basis of azide). HPLC Purity ~ 98%.
M.P.: 178-179oC; MS : 414 (M+1); M.F.: C19H25F2N3O5.
Method B:
A solution of (5R)-3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)-phenyl]-5-azidomethyl-oxazolidin-2-one (50 g, 0.125 mol) in ethyl acetatel (1L ml), were charged with 5g of 10% of Pd-C catalyst(50% wet) and the resulting mixture was hydrogenated at 30psi for 3h at 50oC.. The resulting mixture was cooled and filtered under suction over celite bed. The residue was washed with additional ethyl acetate (200ml). The combined filtrates were concentrated to 500ml volume.
To the above ethyl acetate solution was added Triethyl amine (19.1g, 0.189 mol), and acetic anhydride (16.1g, 1.58mol) in a single lot in few minutes). The reaction mixture was stirred for 16h at R.T. .The resulting mixture was cooled to 0-5oC, stirred for 0.5h and filtered under suction. The residue was washed with cold ethyl acetate(100ml) and dried at 70oC under reduced pressure to obtain the product as a a off-white solid, 43.5g, in 84% yield over two steps.
HPLC Purity ~ 98%
M.P.: 178-179oC; MS : 414 (M+1); M.F.: C19H25F2N3O5.
Method C:
To a solution of (S)-N-2-{3-[3,5-Difluoro-4-(4-methoxymethyl-4-hydroxypiperidine-1yl)phenyl]-2-oxo-oxazolidin-5-yl methyl}-phthalimide (2.77g, 0.0055mol) in ethanol (20ml) was added hydrazine hydrate ( 0.554g, 0.011mol) and the resulting solution stirred at RT for 6h. The solvent was evaporated under reduced pressure, the residue suspended in 3% sodium carbonate solution and extracted in dichloromethane (40ml). The dichloromethane layer was dried and to this solution was added triethylamine(1.11g, 0.011mol) and acetic anhydride (0.67g, 0.007mol) and the solution stirred for 6h at RT. The solvent was evaporated under reduced pressure and the residue purified by flash chromatography to obtain the product as white solid, 1.94g, in 85% yield.
M.P.: 178-179oC; MS: 414 (M+1); M.F.: C19H25F2N3O5.
Method D:
A mixture of (5R)-{3-[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-1-yl)phenyl]-2-oxo-oxazolidin-5-yl methyl}-methanesulfonate (1gm, 4.4mmol) and sodium diformylamide (2gms, 22mmol) in DMF (5ml) was stirred at 95 ºC. for 15hrs. Then a mixture of conc. HCl (0.6ml) and water (0.6ml) and ethanol (8ml) were added. The solution was stirred at 75ºC for 5hrs. The mixture was concentrated under reduced pressure at 60-75 ºC. Water (1ml), ammonia solution (0.5ml) and acetic anhydride (1ml) was added to the residue and the mixture stirred at 70-75 ºC for 4-5 hrs. The solution was cooled to room temperature, diluted with water (5ml) and the separated solid filtered. The residue was washed with water (4ml.) and dried in a vacuum oven at 50ºC to obtain the product as an off-white solid, 0.37g, in 41% yield.
M.P.: 178-179oC; MS : 414 (M+1); M.F.: C19H25F2N3O5.
Method E:
To tetrahydrofuran (30 ml) were added triphenylphosphine (2.11g, 8 mmol)) and diethyldiazocarboxylate (1.62g, 8 mmol)), and the solution stirred at room temperature. After 10 min acetamide (0.475g, 8 mmol)) was added and after a further stirring for 10 min, (R)-3-(3,5-difluoro-4-(4-hydroxy-4-(methoxymethyl)piperidin-1-yl)phenyl)-5-(hydroxymethyl) oxazolidin-2-one (2g, 5.3 mmol) was added and stirring continued further at room temperature. After 16 hrs ice-cold water (4ml) was added to the reaction mixture and the resulting mixture was extracted by ethyl acetate (2 x 20ml). The ethyl acetate extract was dried (over sodium sulfate) and concentrated under reduced pressure. The residue was chromatographed on a column of silica gel to obtain the product as an off-white solid, 0.50g, yield 22%.
M.P.: 178-179oC; MS: 414 (M+1); M.F.: C19H25F2N3O5.
Preparation of Intermediate -11: (S)-N-{3-[3,5-Difluoro-4-(4-methoxymethyl-4-di-O-benzylphosphoryloxy-piperi din-1yl)-phenyl]-2-oxo-oxazolidin-5-ylmethyl}-acetamide
To a solution of (S)-N-{3-[3,5-difluoro-4-(4-methoxymethyl-4-hydroxypiperidine-1yl)-phenyl]-2-oxo-oxazolidin-5-yl methyl}-acetamide (0.2 mmol) and tetrazole (0.6 mmol) in dichloromethane (5 ml) was added dibenzyl N,N,diisopropylphosphoramidite (0.4 mmol) and the resulting mixture was stirred for 4h. The resulting solution was cooled to 0 oC and 0.6 ml of 0.5M m-chloroperbenzoic acid solution in dichloromethane was added. After 4h, the solvent was evaporated under residue pressure and the residue chromatographed on a column of silica gel to obtain the product as a off-white solid in 75% yield,
MS: 674 (M+1); M.F. C33H38F2N3O8P;
Example A: Phosphoric acid mono-(1-{4-[(S)-5-(acetylamino-methyl)-2-oxo-oxazolidin-3-yl]-2,6-difluorophenyl}-4-methoxymethyl-piperidin-4-yl) ester
To a suspension of (S)-N-{3-[3,5-difluoro-4-(4-methoxymethyl-4-di-O-benzylphosphoryl- oxypiperidine-1yl)phenyl]-2-oxo-oxazolidin-5-yl methyl}-acetamide (0.15 mmol) and 20 % palladium hydroxide (20 mg) in 20 ml of a mixture of dichloromethane /aqueous methanol was stirred at room temperature for 6h. The catalyst was filtered and the residue evaporated under reduced pressure. The residue obtained was triturated with acetone to obtain a white solid as product in 70% yield.
MP. >140 °C; MS : 494(M+1) M.F.: C19H26F2N3O8P.
PATENT
WO 2012059823http://www.google.co.in/patents/WO2012059823A1?cl=en
Phosphoric acid mono-(l-{4-[(S)-5-(acetylamino-
methyl)-2-oxo-oxazolidin-3-yl]-2,6-difluorophenyl}-4-methoxymethyl-piperidin-4-yl)
ester of Formula (A),
the process comprising the steps of:
a) Converting intermediate of Formula (1) into intermediate of Formula (3)

b) Converting intermediate of Formula (3) into intermediate of Formula (5)


(5) <6> d) Converting intermediate of Formula (6) into intermediate of Formula (10)

e) Converting intermediate of Formula (10) into intermediate of Formula (11),




Scheme-1
Preparation of Intermediate 10: (5S)- N-{ 3-[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl- piperidin- 1 -yl)-phenyl] -2-oxo-oxazolidin-5-ylmethyl } -acetamide
via
Intermediate 9: 5-aminomethyl-3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-l- yl)-phenyl] -oxazolidin-2-one
Method A:
To a solution of (5R)-3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-l-yl)- phenyl]-5-azidomethyl-oxazolidin-2-one (10 g, 0.025 mol) in methanol (100 ml), were charged cobalt chloride (0.6 g, 0.0025 mol) followed by sodium borohydride (0.95 g, 0.025 mol) in small lots over a period of 30 minutes. The reaction mixture was stirred at RT for additional 2 h. After completion of the reaction , the contents were evaporated under vacuum below 40°C to obtain a sticky mass. The contents were suspended in a mixture of water (100 ml) and ethyl acetate (50 ml) and stirred for 15 minutes. The contents were filtered through a filter-aid bed and the bed was washed with ethyl acetate (2 X 25 ml). The layers were separated and the aqueous layer was further extracted with ethyl acetate (4 X 50 ml). The combined organic layer was washed with 1% HC1 solution (100 ml). The aqueous layer was separated and washed with dichloromethane (4 X 50 ml). The pH of the aqueous layer was adjusted to 8 by adding saturated sodium bicarbonate solution. The contents were extracted with ethyl acetate (6 X 50 ml) till no amine spot was seen in the final organic extract. The combined organic layer (containing the intermediate 5-aminomethyl-3-[3,5-difluoro-4-(4- hydroxy-4-methoxymethyl-piperidin-l-yl)-phenyl]-oxazolidin-2-one) was dried over anhydrous sodium sulfate.
Triethylamine (3.3 g, 4.5 ml, 0.0327 mol) was added to the above organic layer and acetyl chloride (2.17 g, 2 ml, 0.0277 mol) was added gradually over a period of 1 h at RT. The reaction mixture was stirred for 2 h and after completion of the reaction (TLC), the contents were washed with water (50 ml) and the layers separated. Activated carbon (1 g) was added to the organic layer and the contents were stirred for 15 minutes. The contents were filtered on a celite bed and the carbon-celite bed was washed with ethyl acetate (2 X 10 ml). The combined filtrate was evaporated under vacuum to obtain a slurry, which was filtered on a Buchner assembly and the product was washed with ethyl acetate (2 X 10 ml). The product was dried under vacuum at 70°C to obtain 5 g off-white solid. Yield = 48% (on the basis of azide). HPLC Purity ~ 98%.
M.P.: 178-179°C; MS : 414 (M+l); M.F.: C19H25F2N3O5. Method B:
A solution of (5R)-3-[3,5-difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-l-yl)-phenyl]-5- azidomethyl-oxazolidin-2-one (50 g, 0.125 mol) in ethyl acetatel (1L ml), were charged with 5g of 10% of Pd-C catalyst(50% wet) and the resulting mixture was hydrogenated at 30psi for 3h at 50°C. The resulting mixture was cooled and filtered under suction over celite bed. The residue was washed with additional ethyl acetate (200ml). The combined filtrates were concentrated to 500ml volume. To the above ethyl acetate solution was added Triethyl amine (19. lg, 0.189 mol), and acetic anhydride (16. lg, 1.58mol) in a single lot in few minutes). The reaction mixture was stirred for 16h at R.T. .The resulting mixture was cooled to 0-5°C, stirred for 0.5h and filtered under suction. The residue was washed with cold ethyl acetate( 100ml) and dried at 70°C under reduced pressure to obtain the product as a a off-white solid, 43.5g, in 84% yield over two steps.
HPLC Purity ~ 98%
M.P.: 178-179°C; MS : 414 (M+l); M.F.: C19H25F2N3O5. Method C:
To a solution of (S)-N-2-{3-[3,5-Difluoro-4-(4-methoxymethyl-4-hydroxypiperidine- lyl)phenyl]-2-oxo-oxazolidin-5-yl methyl }-phthalimide (2.77g, 0.0055mol) in ethanol (20ml) was added hydrazine hydrate ( 0.554g, 0.01 lmol) and the resulting solution stirred at RT for 6h. The solvent was evaporated under reduced pressure, the residue suspended in 3% sodium carbonate solution and extracted in dichloromethane (40ml). The dichloromethane layer was dried and to this solution was added triethylamine(l.l lg, 0.01 lmol) and acetic anhydride (0.67g, 0.007mol) and the solution stirred for 6h at RT. The solvent was evaporated under reduced pressure and the residue purified by flash chromatography to obtain the product as white solid, 1.94g, in 85% yield.
M.P.: 178-179°C; MS: 414 (M+l); M.F.: C19H25F2N3O5. Method D:
A mixture of (5R)-{3-[3,5-Difluoro-4-(4-hydroxy-4-methoxymethyl-piperidin-l-yl)phenyl]- 2-oxo-oxazolidin-5-yl methyl }-methanesulfonate (lgm, 4.4mmol) and sodium diformylamide (2gms, 22mmol) in DMF (5ml) was stirred at 95 °C. for 15hrs. Then a mixture of cone. HC1 (0.6ml) and water (0.6ml) and ethanol (8ml) were added. The solution was stirred at 75°C for 5hrs. The mixture was concentrated under reduced pressure at 60-75 °C. Water (1ml), ammonia solution (0.5ml) and acetic anhydride (1ml) was added to the residue and the mixture stirred at 70-75 °C for 4-5 hrs. The solution was cooled to room temperature, diluted with water (5ml) and the separated solid filtered. The residue was washed with water (4ml.) and dried in a vacuum oven at 50°C to obtain the product as an off-white solid, 0.37g, in 41% yield.
M.P.: 178-179°C; MS : 414 (M+l); M.F.: C19H25F2N3O5. Method E:
To tetrahydrofuran (30 ml) were added triphenylphosphine (2.1 lg, 8 mmol)) and diethyldiazocarboxylate (1.62g, 8 mmol)), and the solution stirred at room temperature. After 10 min acetamide (0.475g, 8 mmol)) was added and after a further stirring for 10 min, (R)-3- (3,5-difluoro-4-(4-hydroxy-4-(methoxymethyl)piperidin-l-yl)phenyl)-5-(hydroxymethyl) oxazolidin-2-one (2g, 5.3 mmol) was added and stirring continued further at room temperature. After 16 hrs ice-cold water (4ml) was added to the reaction mixture and the resulting mixture was extracted by ethyl acetate (2 x 20ml). The ethyl acetate extract was dried (over sodium sulfate) and concentrated under reduced pressure. The residue was chromatographed on a column of silica gel to obtain the product as an off-white solid, 0.50g, yield 22%.
M.P.: 178-179°C; MS: 414 (M+l); M.F.: C19H25F2N3O5.
PATENT
http://www.google.co.in/patents/WO2008038092A2?cl=enWockhardt Research Center,


‘ Scheme-1 ‘
/////////
DR ANTHONY MELVIN CRASTO Ph.D
GLENMARK SCIENTIST , NAVIMUMBAI, INDIA
did you feel happy, a head to toe paralysed man's soul in action for you round the clock
need help, email or call me
MOBILE-+91 9323115463
web link
I was paralysed in dec2007WILL BE UPDATED
EperezolidradezolidRanbezolid
Sutezolidlinezolid
I appreciate you considering me for this position. I look forward to hearing from you. Thank you for sharing your opinion. I value your honesty and will respond as quickly as possible.
ReplyDelete