
HERE
TINIB SERIES 1/3
1 IBRUTINIB
2.COBIMETINIB
3.AFATINIB
4 SELUMETINIB
5 ALECTINIB
6 IMATINIB
7 NERATINIB
8 ERLOTINIB
9 AMUVATINIB
SEE
http://drugsynthesisint.blogspot.in/p/tinib-series23.html SET 2/3
10 BINIMETINIB
11 DACOMITINIB
12 MOMELOTINIB
13 PALBOCICLIB
14 SORAFENIB
15 NINTEDANIB
16 LAPATINIB
17 QUIZARTINIB
18 BOSUTINIB
19
SET 1/3 STARTS HERE
1 IBRUTINIB

 IBRUTINIB
IBRUTINIB















2 Cobimetinib


Cobimetinib
934660-93-2 cas ………….(S )enantiomer desired
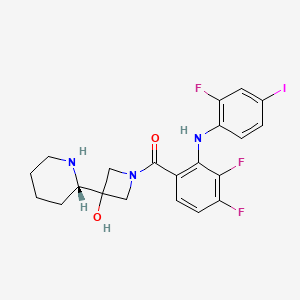
[3,4-Difluoro-2-(2-fluoro-4-iodoanilino)phenyl]{3-hydroxy-3-[(2S)-piperidin-2-yl]azetidin-1-yl} methanone
l-({3,4-difluoro-2-[(2-fluoro-4-iodophenyl)amino]phenyl}carbonyl)-3-[(2S)-piperidin-2- yl]azetidin-3-ol
1-[3,4-Difluoro-2-(2-fluoro-4-iodophenylamino)phenyl]-1-[3-hydroxy-3-[2(S)-piperidinyl]azetidin-1-yl]methanone
Cobimetinib, XL518, GDC 0973, 934660-93-2, XL 518, GDC0973
Molecular Formula: C21H21F3IN3O2
Molecular Weig,ht: 531.31002
other isomer and racemate
 Cobimetinib (racemate)
Cobimetinib (racemate)
CAS No: 934662-91-6
 Cobimetinib (R-enantiomer)
Cobimetinib (R-enantiomer)CAS No: 934660-94-3
 cobimetinib fumarate [USAN]
cobimetinib fumarate [USAN]
Molecular Formula: C46H46F6I2N6O8
Average mass: 1178.692261 Da
Average mass: 1178.692261 Da
(2E)-2-Butendisäure –{3,4-difluor-2-[(2-fluor-4-iodphenyl)amino]phenyl}{3-hydroxy-3-[(2S)-2-piperidinyl]-1-azetidinyl}methanon (1:2) [German] [ACD/IUPAC Name]
{3,4-Difluoro-2-[(2-fluoro-4-iodophenyl)amino]phenyl}{3-hydroxy-3-[(2S)-2-piperidinyl]-1-azetidinyl}methanone (2E)-2-butenedioate (2:1) [ACD/IUPAC Name]
1369665-02-0 [RN]
Acide (2E)-2-butènedioïque - {3,4-difluoro-2-[(2-fluoro-4-iodophényl)amino]phényl}{3-hydroxy-3-[(2S)-2-pipéridinyl]-1-azétidinyl}méthanone (1:2) [French][ACD/IUPAC Name]
Bis({3,4-difluoro-2-[(2-fluoro-4-iodophenyl)amino]phenyl}{3-hydroxy-3-[(2S)-piperidin-2-yl]azetidin-1-yl}methanone) (2E)-but-2-enedioate
Methanone, [3,4-difluoro-2-[(2-fluoro-4-iodophenyl)amino]phenyl][3-hydroxy-3-[(2S)-2-piperidinyl]-1-azetidinyl]-, (2E)-2-butenedioate (2:1) (salt
Cobimetinib (GDC-0973, XL-518) is a MEK inhibitor being developed by Exelixis and Roche. It is being studied in combination withvemurafenib to treat several cancers, including melanoma.
Cobimetinib is an inhibitor of MEK kinase, which is an enzyme that the mitogen-activated protein kinase (MAPK). This compound was originated by Exelixis and is being developed by Genentech. Currently, Cobimetinb is in phase III trials at Genentech for the treatment of metastatic melanoma and in phase I clinical trials for the treatment of solid tumors. Cobimetinib has received an orphan drug designation in the U.S. for treatment of stage IIb, IIc, III, and IV melanoma with BRAFV600E mutation.
GDC-0973 (XL-518; GDC 0973) is a selective inhibitor of MEK GDC-0973 is also known as mitogen activated protein kinase kinase (MAPKK), is a key component of the RAS / RAF / MEK / ERK pathway, which. is frequently activated in human tumors.
Inappropriate activation of the MEK / ERK pathway promotes cell growth in the absence of exogenous growth factors.
The ERK/MAP kinase cascade is a key mechanism subject to dysregulation in cancer and is constitutively activated or highly upregulated in many tumor types. Mutations associated with upstream pathway components RAS and Raf occur frequently and contribute to the oncogenic phenotype through activation of MEK and then ERK. Inhibitors of MEK have been shown to effectively block upregulated ERK/MAPK signaling in a range of cancer cell lines and have further demonstrated early evidence of efficacy in the clinic for the treatment of cancer. Guided by structural insight, a strategy aimed at the identification of an optimal diphenylamine-based MEK inhibitor with an improved metabolism and safety profile versus PD-0325901 led to the discovery of development candidate 1-({3,4-difluoro-2-[(2-fluoro-4-iodophenyl)amino]phenyl}carbonyl)-3-[(2S)-piperidin-2-yl]azetidin-3-ol (XL518, GDC-0973) (1). XL518 exhibits robust in vitro and in vivo potency and efficacy in preclinical models with sustained duration of action and is currently in early stage clinical trials.
Process for the preparation of MEK inhibitors, relates cobimetinib. Genentech and its parent company Roche, under license from Exelixis, are developing cobimetinib, for the treatment of solid tumors, including melanoma, which is in phase 3 trials as of April 2014. The drug was originally disclosed in WO2007044515. For a previous filing on MEK inhibitors, see WO2008124085.
patent
EXAMPLE 22(a) and 22(b) l-({3,4-difluoro-2-[(2-fluoro-4-iodophenyϊ)amino]phenyl}carbonyI)-3-[(2R)-piperidin-2- yl]azetidin-3-ol

l-({3,4-difluoro-2-[(2-fluoro-4-iodophenyl)amino]phenyl}carbonyl)-3-[(2S)-piperidin-2- yl]azetidin-3-ol
 desired
desired
[00334] To a solution of 1 , 1 -dimethylethyl 2-(3 -hydroxy- 1 -
{[(phenylmethyl)oxy]carbonyl}azetidin-3-yl)piperidine-l-carboxylate (368 mg, 0.94 mmol), prepared using procedures similar to those described in Reference 5, in dichloromethane (5 mL) was added DMAP (115 mg, 0.94 mmol) and the resulting solution was cooled to O0C. (i?)-(-)-α-Methoxy-α-trifluoromethylphenylacetyl chloride (105 μL, 0.56 mmol) was added to the solution by syringe and the mixture was allowed to warm to room temperature then stirred an additional 12 hours. The solution was then partitioned with saturated aqueous soldium bicarbonate and the organic phase dried over anhydrous magnesium sulfate then filtered and concentrated to an oily residue.
Silica gel flash chromatography using hexanes:ethyl acetate 3:1 as eluent afforded the less polar 1,1 -dimethyl ethyl (2R)-2-(l- {[(phenylmethyl)oxy]carbonyl}-3-{[(2i?)-3,3,3-trifluoro-2-(methyloxy)-2- phenylpropanoyl]oxy}azetidin-3-yl)piperidine-l-carboxylate (27.5 mg, 5% yield), the more polar 1 , 1 -dimethylethyl (2S)-2-(l -{ [(phenylmethyl)oxy]carbonyl} -3-{ [(2i?)-3,3,3-trifluoro-2- (methyloxy)-2-phenylpropanoyl]oxy}azetidin-3-yl)piperidine-l-carboxylate (105 mg, 19% yield) and starting material (253 mg, 69% recovery).
[00335] The starting material thus recovered was taken into dichloromethane (3 mL) followed by addition of DMAP (115 mg, 0.94 mmol) and (i?)-(-)-α-methoxy-α- trifluoromethylphenylacetyl chloride (105 μL, 0.56 mmol) and the mixture was allowed to stir at room temperature over 12 hours. Proceeding as before afforded combined 1,1- dimethylethyl (2R)-2-(l-{[(phenylmethyl)oxy]carbonyl}-3-{[(2i?)-3,3,3-trifluoro- 2-(methyloxy)-2-phenylpropanoyl]oxy}azetidin-3-yl)piperidine-l-carboxylate (46.6 mg, 8% yield), the more polar 1,1 -dimethylethyl (25)-2-(l-{[(phenylmethyl)oxy]carbonyl}-3-{[(2i?)- 3,3,3-trifluoro-2-(methyloxy)-2-phenylpropanoyl]oxy}azetidin-3-yl)piperidine-l-carboxylate (228 mg, 41% yield) and starting material (100.8 mg, 27% recovery). [00336] The starting material thus recovered was taken into tetrahydrofuran: dichloromethane (1 :1, 2 mL) followed by addition of DMAP (47 mg, 0.39 mmol) and (R)-(-)-α-methoxy-α-trifluoromethylphenylacetyl chloride (80 μL, 0.43 mmol) and the mixture was heated to 60 0C over 12 hours.
Proceeding as before afforded combined less polar 1,1-dimethylethyl (2i?)-2-(l-{[(phenylmethyl)oxy]carbonyl}-3-{[(2i?)-3,3,3- trifluoro-2-(methyloxy)-2-phenylpropanoyl] oxy } azetidin-3 -yl)piperidine- 1 -carboxylate ( 144 mg, 26 % yield). The chiral ester derivatives thus obtained were again subject to silica gel flash chromatography using hexanes:ethyl acetate 3:1 as eluent to give the pure less polar 1,1-dimethylethyl (2i?)-2-(l-{[(phenylmethyl)oxy]carbonyl}-3-{[(2i?)-3,3,3-trifluoro-2-
(methyloxy)-2-phenylpropanoyl]oxy}azetidin-3-yl)piperidine-l-carboxylate (122.8 mg, 22% yield) and the more polar 1,1-dimethylethyl (2£)-2-(l-{[(phenylmethyl)oxy]carbonyl}-3- { [(2R)-3 ,3 ,3 -trifluoro-2-(methyloxy)-2-phenylpropanoyl] oxy } azetidin-3-yl)piperidine- 1 – carboxylate {111.6 mg, 32% yield) both as colorless amorphous residues. [00337] l,l-Dimethylethyl (2i?)-2-(l-{[(phenylmethyl)oxy]carbonyl}-3-{[(2i?)-3,3,3- trifluoro-2-(methyloxy)-2-phenylρropanoyl]oxy}azetidin-3-yl)piperidine-l-carboxylate (122.8 mg, 0.21 mmol) was taken into methanol (4 mL) followed by addition of IM aqueous sodium hydroxide (1 mL) and the resulting solution was stirred for one hour at room temperature.
The solution was then partitioned with ethyl acetate and IN aqueous hydrochloric acid. The organic layer was washed with brine, dried over anhydrous magnesium sulfate then filtered and concentrated. The residue was purified by silica gel flash chromatography using hexanes:ethyl acetate 2:1 to give 1,1-dimethylethyl (2i?)-2-(3- hydroxy- 1 – { [(phenylmethyl)oxy] carbonyl } azetidin-3 -yl)piperidine- 1 -carboxylate (60.8 mg, 81% yield) a colorless amorphous solid. 1,1-dimethylethyl (2ιS)-2-(3-hydroxy-l- {[(phenylmethyl)oxy]carbonyl}azetidin-3-yl)piperidine-l-carboxylate (87.4 mg, 75% yield) was prepared analogously.
[00338] 1 , 1 -Dimethylethyl (2i?)-2-(3-hydroxy- 1 – { [(phenylmethyl)oxy] carbonyl } azetidin- 3 -yl)piperidine-l -carboxylate (60.8 mg, 0.16 mmol) and 10% Pd/C (30 mg) were taken into methanol (2 mL) and the mixture hydrogenated at ambient pressure for one hour. The suspension was then filtered through a celite pad and concentrated then dried in vacuo to a colorless solid. The solid amine was taken into THF (1 mL) followed by addition of DIPEA (42 μL, 0.24 mmol) and 3,4-difluoro-2-[(2-fluoro-4-iodophenyl)amino]benzoyl fluoride (63 mg, 0.16 mmol), prepared using procedures similar to those described in Reference 1, and the mixture stirred at room temperature for 30 minutes.
The reaction mixture was partitioned with ethyl acetate and 1 N aqueous hydrochloric acid and the organic layer washed with brine, dried over anhydrous magnesium sulfate then filtered and concentrated. Purification of the residue by silica gel flash chromatography using hexanes: ethyl acetate 3:2 as eluent afforded 1,1-dimethylethyl (2i?)-2-[l-({3,4-difluoro-2-[(2-fluoro-4- iodophenyl)amino]phenyl}carbonyl)-3-hydroxyazetidin-3-yl]piperidine-l -carboxylate (74.9 mg, 74% yield) as an amorphous solid. 1,1 -Dimethylethyl (2i?)-2-[l-({3,4-difluoro-2-[(2- fluoro~4-iodophenyl)amino]phenyl}carbonyl)-3-hydroxyazetidin-3-yl]piperidine-l- carboxylate 1R NMR (400 MHz, CDCl3): 8.53 (br s, 0.5H), 8.40 (br s, 0.5H), 7.41-7.38 (dd, IH), 7.34-7.31(dt, IH), 7.17-7.14 (m, IH), 6.86-6.79 (m, IH), 6.63-6.587 (m, IH), 4.24-3.90 (m, 4H), 3.37-3.23 (m, IH), 2.90-2.80 (m, IH), 1.85-1.54 (m, 7H), 1.43 (s, 9H); MS (EI) for C26H29F3IN3O4: 576 (M-C4H9 4).
[00339] 1 , 1 -dimethylethyl (2R)-2-[l -({3,4-difluoro-2-[(2-fluoro-4- iodopheny^aminojphenylJcarbonyO-S-hydroxyazetidin-S-yljpiperidine-l-carboxylate (74.9 mg, 0.12 mmol) was taken into methanol (1 mL) followed by addition of 4 N HCl in dioxane (1 mL) and the solution was stirred at room temperature for one hour. The solution was then concentrated and the residue partitioned with chloroform and saturated aqueous sodium bicarbonate.
The organic layer was washed with brine, dried over anhydrous sodium' sulfate then filtered and concentrated. Purification of the residue by silica gel flash chromatography using ethyl acetate then concentrated aqueous ammonia in chloroform and methanol (0.1 :10:1) as eluents afforded l-({3,4-difluoro-2-[(2-fluoro-4- iodophenyl)amino]phenyl}carbonyl)-3-[(2i?)-piperidin-2-yl]azetidin-3-ol (57.3 mg) as a colorless amorphous solid.
The free base was taken into methanol (1 mL) then brought to about pH 1 by addition of 4 N HCl in dioxane and the solution concentrated. The residue was triturated with ethyl ether to afford a suspension. The solid was collected by filtration to afford l-({3,4-difluoro-2-[(2-fluoro-4-iodophenyl)amino]phenyl}carbonyl)-3-[(2i?)- piperidin-2-yl]azetidin-3-ol hydrochloride salt (49 mg, 72% yield) as a colorless solid. 1H NMR (400 MHz, CDCl3): 8.43-8.39 (d, IH), 7.41-7.38 (dd, IH), 7.33-7.31(dt, IH), 7.14- 7.10 (m, IH), 6.84-6.80 (m, IH), 6.63-6.57 (m, IH), 4.12-3.99 (m, 4H), 3.10-3.08 (d, IH), 2.72-2.69 (d, IH), 2.64-2.62 (m, IH), 1.61-1.58 (m, 2H), 1.36-1.16 (m, 4H); MS (EI) for C21H2IF3IN3O2: 532 (MH+).

………………….
[3,4-difluoro-2-[(2-fluoro-4- iodophenyl)amino]phenyl][3-hydroxy-3-[(2S)-2-piperidinyl]-l-azetidinyl]methanone.
GDC-0973 has the chemical structure:

[00156] Compound II may be prepared following the methods described in
US2009/0156576 (the contents of which are hereby incorporated by reference). Compound II has the following CAS Registry Number: 934660-93-2 .
Example 22(a) and 22(b) 1-({3,4-difluoro-2-[(2-fluoro-4-iodophenyl)amino]phenyl}carbonyl)-3-[(2R)-piperidin-2-yl]azetidin-3-ol

and 1-({3,4-difluoro-2-[(2-fluoro-4-iodophenyl)amino]phenyl}carbonyl)-3-[(2S)-piperidin-2-yl]azetidin-3-ol

To a solution of 1,1-dimethylethyl 2-(3-hydroxy-1-{[(phenylmethyl)oxy]carbonyl}azetidin-3-yl)piperidine-1-carboxylate (368 mg, 0.94 mmol), prepared using procedures similar to those described in Reference 5, in dichloromethane (5 mL) was added DMAP (115 mg, 0.94 mmol) and the resulting solution was cooled to 0° C. (R)-(−)-α-Methoxy-α-trifluoromethylphenylacetyl chloride (105 μL, 0.56 mmol) was added to the solution by syringe and the mixture was allowed to warm to room temperature then stirred an additional 12 hours. The solution was then partitioned with saturated aqueous sodium bicarbonate and the organic phase dried over anhydrous magnesium sulfate then filtered and concentrated to an oily residue. Silica gel flash chromatography using hexanes:ethyl acetate 3:1 as eluent afforded the less polar 1,1-dimethylethyl (2R)-2-(1-{[(phenylmethyl)oxy]carbonyl}-3-{[(2R)-3,3,3-trifluoro-2-(methyloxy)-2-phenylpropanoyl]oxy}azetidin-3-yl)piperidine-1-carboxylate (27.5 mg, 5% yield), the more polar 1,1-dimethylethyl (2S)-2-(1-{[(phenylmethyl)oxy]carbonyl}-3-{[(2R)-3,3,3-trifluoro-2-(methyloxy)-2-phenylpropanoyl]oxy}azetidin-3-yl)piperidine-1-carboxylate (105 mg, 19% yield) and starting material (253 mg, 69% recovery).
The starting material thus recovered was taken into dichloromethane (3 mL) followed by addition of DMAP (115 mg, 0.94 mmol) and (R)-(−)-α-methoxy-α-trifluoromethylphenylacetyl chloride (105 μL, 0.56 mmol) and the mixture was allowed to stir at room temperature over 12 hours. Proceeding as before afforded combined 1,1-dimethylethyl (2R)-2-(1-{[(phenylmethyl)oxy]carbonyl}-3-{[(2R)-3,3,3-trifluoro-2-(methyloxy)-2-phenylpropanoyl]oxy}azetidin-3-yl)piperidine-1-carboxylate (46.6 mg, 8% yield), the more polar 1,1-dimethylethyl (2S)-2-(1-{[(phenylmethyl)oxy]carbonyl}-3-{[(2R)-3,3,3-trifluoro-2-(methyloxy)-2-phenylpropanoyl]oxy}azetidin-3-yl)piperidine-1-carboxylate (228 mg, 41% yield) and starting material (100.8 mg, 27% recovery).
The starting material thus recovered was taken into tetrahydrofuran:dichloromethane (1:1, 2 mL) followed by addition of DMAP (47 mg, 0.39 mmol) and (R)-(−)-α-methoxy-α-trifluoromethylphenylacetyl chloride (80 μL, 0.43 mmol) and the mixture was heated to 60° C. over 12 hours. Proceeding as before afforded combined less polar 1,1-dimethylethyl (2R)-2-(1-{[(phenylmethyl)oxy]carbonyl}-3-{[(2R)-3,3,3-trifluoro-2-(methyloxy)-2-phenylpropanoyl]oxy}azetidin-3-yl)piperidine-1-carboxylate (144 mg, 26% yield). The chiral ester derivatives thus obtained were again subject to silica gel flash chromatography using hexanes:ethyl acetate 3:1 as eluent to give the pure less polar 1,1-dimethylethyl (2R)-2-(1-{[(phenylmethyl)oxy]carbonyl}-3-{[(2R)-3,3,3-trifluoro-2-(methyloxy)-2-phenylpropanoyl]oxy}azetidin-3-yl)piperidine-1-carboxylate (122.8 mg, 22% yield) and the more polar 1,1-dimethylethyl (2S)-2-(1-{[(phenylmethyl)oxy]carbonyl}-3-{[(2R)-3,3,3-trifluoro-2-(methyloxy)-2-phenylpropanoyl]oxy}azetidin-3-yl)piperidine-1-carboxylate (177.6 mg, 32% yield) both as colorless amorphous residues.
1,1-Dimethylethyl (2R)-2-(1-{[(phenylmethyl)oxy]carbonyl}-3-{[(2R)-3,3,3-trifluoro-2-(methyloxy)-2-phenylpropanoyl]oxy}azetidin-3-yl)piperidine-1-carboxylate (122.8 mg, 0.21 mmol) was taken into methanol (4 mL) followed by addition of 1M aqueous sodium hydroxide (1 mL) and the resulting solution was stirred for one hour at room temperature. The solution was then partitioned with ethyl acetate and 1N aqueous hydrochloric acid. The organic layer was washed with brine, dried over anhydrous magnesium sulfate then filtered and concentrated. The residue was purified by silica gel flash chromatography using hexanes:ethyl acetate 2:1 to give 1,1-dimethylethyl (2R)-2-(3-hydroxy-1-{[(phenylmethyl)oxy]carbonyl}azetidin-3-yl)piperidine-1-carboxylate (60.8 mg, 81% yield) a colorless amorphous solid. 1,1-dimethylethyl (2S)-2-(3-hydroxy-1-{[(phenylmethyl)oxy]carbonyl}azetidin-3-yl)piperidine-1-carboxylate (87.4 mg, 75% yield) was prepared analogously.
1,1-Dimethylethyl (2R)-2-(3-hydroxy-1-{[(phenylmethyl)oxy]carbonyl}azetidin-3-yl)piperidine-1-carboxylate (60.8 mg, 0.16 mmol) and 10% Pd/C (30 mg) were taken into methanol (2 mL) and the mixture hydrogenated at ambient pressure for one hour. The suspension was then filtered through a celite pad and concentrated then dried in vacuo to a colorless solid. The solid amine was taken into THF (1 mL) followed by addition of DIPEA (42 μL, 0.24 mmol) and 3,4-difluoro-2-[(2-fluoro-4-iodophenyl)amino]benzoyl fluoride (63 mg, 0.16 mmol), prepared using procedures similar to those described in Reference 1, and the mixture stirred at room temperature for 30 minutes. The reaction mixture was partitioned with ethyl acetate and 1 N aqueous hydrochloric acid and the organic layer washed with brine, dried over anhydrous magnesium sulfate then filtered and concentrated. Purification of the residue by silica gel flash chromatography using hexanes:ethyl acetate 3:2 as eluent afforded 1,1-dimethylethyl (2R)-2-[1-({3,4-difluoro-2-[(2-fluoro-4-iodophenyl)amino]phenyl}carbonyl)-3-hydroxyazetidin-3-yl]piperidine-1-carboxylate (74.9 mg, 74% yield) as an amorphous solid. 1,1-Dimethylethyl (2R)-2-[1-({3,4-difluoro-2-[(2-fluoro-4-iodophenyl)amino]phenyl}carbonyl)-3-hydroxyazetidin-3-yl]piperidine-1-carboxylate 1H NMR (400 MHz, CDCl3): 8.53 (br s, 0.5H), 8.40 (br s, 0.5H), 7.41-7.38 (dd, 1H), 7.34-7.31 (dt, 1H), 7.17-7.14 (m, 1H), 6.86-6.79 (m, 1H), 6.63-6.587 (m, 1H), 4.24-3.90 (m, 4H), 3.37-3.23 (m, 1H), 2.90-2.80 (m, 1H), 1.85-1.54 (m, 7H), 1.43 (s, 9H); MS (EI) for C26H29F3IN3O4: 576 M-C4H9 +).
1,1-dimethylethyl (2R)-2-[1-({3,4-difluoro-2-[(2-fluoro-4-iodophenyl)amino]phenyl}carbonyl)-3-hydroxyazetidin-3-yl]piperidine-1-carboxylate (74.9 mg, 0.12 mmol) was taken into methanol (1 mL) followed by addition of 4 N HCl in dioxane (1 mL) and the solution was stirred at room temperature for one hour. The solution was then concentrated and the residue partitioned with chloroform and saturated aqueous sodium bicarbonate. The organic layer was washed with brine, dried over anhydrous sodium sulfate then filtered and concentrated. Purification of the residue by silica gel flash chromatography using ethyl acetate then concentrated aqueous ammonia in chloroform and methanol (0.1:10:1) as eluents afforded 1-({3,4-difluoro-2-[(2-fluoro-4-iodophenyl)amino]phenyl}carbonyl)-3-[(2R)-piperidin-2-yl]azetidin-3-ol (57.3 mg) as a colorless amorphous solid. The free base was taken into methanol (1 mL) then brought to about pH 1 by addition of 4 N HCl in dioxane and the solution concentrated. The residue was triturated with ethyl ether to afford a suspension. The solid was collected by filtration to afford 1-({3,4-difluoro-2-[(2-fluoro-4-iodophenyl)amino]phenyl}carbonyl)-3-[(2R)-piperidin-2-yl]azetidin-3-ol hydrochloride salt (49 mg, 72% yield) as a colorless solid. 1H NMR (400 MHz, CDCl3): 8.43-8.39 (d, 1H), 7.41-7.38 (dd, 1H), 7.33-7.31 (dt, 1H), 7.14-7.10 (m, 1H), 6.84-6.80 (m, 1H), 6.63-6.57 (m, 1H), 4.12-3.99 (m, 4H), 3.10-3.08 (d, 1H), 2.72-2.69 (d, 1H), 2.64-2.62 (m, 1H), 1.61-1.58 (m, 2H), 1.36-1.16 (m, 4H); MS (EI) for C21H21F3IN3O2: 532 (MH+).
………………………………..
Novel carboxamide-based allosteric MEK inhibitors: Discovery and optimization efforts toward XL518 (GDC-0973)
ACS Med Chem Lett 2012, 3(5): 416
ACS Med Chem Lett 2012, 3(5): 416
8-17-2011
|
Methods of using MEK inhibitors
| |
3-30-2011
|
Azetidines as MEK Inhibitors for the Treatment of Proliferative Diseases
| |
9-29-2010
|
Azetidines as MEK Inhibitors for the Treatment of Proliferative Diseases
| |
3-26-2010
|
Methods of Using PI3K and MEK Modulators
|
//////////////////////////////
3 AFATINIB

Afatinib
439081-18-2
850140-73-7 dimaleate
Tovok, BIBW2992, Tomtovok
An irreversible EGFR/HER2 inhibitor
| Molecular Weight: | 485.94 |
| Molecular Formula: | C24H25ClFN5O3 |
N-[4-[(3-Chloro-4-fluorophenyl)amino]-7-[[(3S)-tetrahydro-3-furanyl]oxy]-6-quinazolinyl]-4(dimethylamino)-2-butenamide
4 - [(3-chloro-4-fluorophenyl) amino] -6 – {[4 - (N, N-dimethylamino)-1-oxo-2-buten-1-yl] – amino} -7 – ((S )-tetrahydrofuran-3-yloxy)-quinazoline
(E)-4-Dimethylamino-but-2-enoic acid {4-(3-chloro-4-fluoro- phenylanimo)-7-[(S)-(tetrahydro-furan-3-yl) oxy]-quinazolin-6-yl} -amide
4 – [(3_ chloro-4 - fluorophenyl) amino] -6 – {[4_ (N, N-dimethylamino)-buten-1-oxo-_2_ - yl] amino}-7 – ((S) – tetrahydrofuran-3 – yloxy) – quinazoline
The endorsement for Giotrif (afatinib) covers the drug’s use in the treatment of adult patients with locally advanced or metastatic non-small cell lung cancer (NSCLC) who have the epidermal growth factor receptor (EGFR) gene mutation, which is present in about 10 per cent of people with NSCLC.
It caps a good month for Boehringer, which won US approval for the drug under the brand name Gilotrif two weeks ago, adding to the company’s list of therapy areas, which so far include chronic obstructive pulmonary disease (COPD), anticoagulation, HIV, Parkinson’s disease and diabetes.
In the US, the drug is approved alongside a companion diagnostic to help determine if a patient’s lung cancer cells express the EGFR mutations, whereas the EMA recommendation just includes the requirement that Giotrif be initiated and supervised by a physician experienced in the use of anti-cancer therapies.
GILOTRIF tablets contain afatinib, a tyrosine kinase inhibitor which is a 4-anilinoquinazoline. Afatinib is presented as the dimaleate salt, with the chemical name 2-butenamide, N-[4-[(3-chloro-4-fluorophenyl)amino]7-[[(3S)-tetrahydro-3-furanyl]oxy]-6-quinazolinyl]-4-(dimethylamino)-,(2E)-, (2Z)-2-butenedioate (1:2). Its structural formula is:
 |
Afatinib dimaleate is a white to brownish yellow powder, water soluble and hygroscopic, with an empirical formula of C32H33ClFN5O11, and a molecular weight of 718.1 g/mol.
GILOTRIF tablets for oral administration are available in 40 mg, 30 mg, or 20 mg of afatinib (equivalent to 59.12 mg, 44.34 mg, or 29.56 mg afatinib dimaleate, respectively). The inactive ingredients of GILOTRIF are the following: Tablet Core: lactose monohydrate, microcrystalline cellulose, crospovidone, colloidal silicon dioxide, magnesium stearate. Coating: hypromellose, polyethylene glycol, titanium dioxide, talc, polysorbate 80, FD&C Blue No. 2 (40 mg and 30 mg tablets only).

Afatinib (BIBW2992) is an irreversible EGFR/Neu inhibitor with an IC50 of 14 nM. Afatinib is a potent inhibitor of EGFR phosphorylation. Afatinib showed positive results in assays against a variety of human cancer cell lines, including A431, murine NIH-3T3 cells, and breast cancer cell line BT-474.
Afatinib[2] (INN; trade name Gilotrif in the US and Giotrif in Europe, previously Tomtovok and Tovok[3]) is a drug approved inmuch of the world (including the United States, Canada, the United Kingdom and Australia) for the treatment of metastatic non-small cell lung carcinoma (NSCLC), developed by Boehringer Ingelheim.[4][5][6] It acts as an angiokinase inhibitor.
Quinazoline derivatives, such as afatinib, are described in WO2002050043. This document also describes certain favourable pharmacological properties of this compound. The dimaleate salt and its crystalline form are described in WO2005037824.
It is known in the W002/50043, which describes the pharmacological properties has important compounds include in particular their pharmacological properties mediated by the tyrosine kinase inhibitory effect and the signal transmission through the skin growth factor receptor (EGF-R) signal transduction mediated inhibitory effect. Therefore, this type of compounds are useful in the treatment of diseases, in particular for the treatment of tumor diseases, lung and gastrointestinal and respiratory tract and gall bladder and bile duct disease.
W002/50043 discloses a method for preparing a compound wherein the amino crotonic group (IV), such as 4_ [(3 - chloro-4 - fluorophenyl) amino] -6 – {[4 - (N, N-two methyl-amino)-oxo-2-1_ - buten-1 - yl] amino} -7 – ((S) – tetrahydrofuran-3 – yloxy) – quinazoline in the one-pot reaction from the corresponding aniline component (II), bromo crotonic acid (III), oxalyl chloride and a secondary amine prepared (see Figure 1).
Figure 1:

In the method, the yield was 50% at most. In addition, the implementation typically purified by column chromatography. Therefore Preparation of 4 – [(3_ chloro-4 - fluorophenyl) amino] -6 – {[4 - (N, N-dimethylamino)-l-oxo-2 - buten-1 - yl] amino} -7 – ((S) – tetrahydrofuran-3 – yloxy) – quinazoline of the method is not for large-scale industrial production. Moreover, the method is not drawback bromo crotonate purchased by a large number of commercial sources, and the corresponding bromo-methyl crotonate only be obtained in a purity of about 80%.These methods are used in this case is also 4 – [(3 - chloro-4 - fluorophenyl) amino] -6 – {[4 - (N, N-dimethylamino) -1 - oxo - butene-1 - yl] amino} -7 – (⑶ – tetrahydrofuran-3 – yloxy) – quinazoline industrialized production adversely affect the applicability.
In the above-mentioned drawbacks of known production methods, the present invention is to provide a produce aminocrotonate aryl amides, in particular 4 – [(3 - chloro-4 - fluorophenyl) amino] -6 – {[4 - (N, N-dimethylamino)-buten-1-oxo-_2_ - yl] amino} -7 – ((S) – tetrahydrofuran-3 – yloxy) – quinazoline The method of the method can be easily obtained using high purity starting materials and does not require the use of any material technology. Thus, the new method should be applicable on an industrial scale synthesis grade and therefore suitable for commercial applications.
This task is according to the present invention for preparing 4 – [(3 - chloro-4 - fluorophenyl) amino] -6 – {[4 - (N, N-dimethylamino) -1 - oxo-2 - buten-1 - yl] amino} -7 – (⑶ – tetrahydrofuran-3 – yloxy) – quinazoline, and other amino crotonic method based compound. In addition to high yield industrially embodiment, the synthesis method according to the present invention also has a very good purity and less than 0.1 of the advantages of a low cis content.
According to Figure 2, in the method according to the present invention, an aryl group corresponding amino compound (V) with two – (Ch-ware yl) _ phosphono acetic acid, preferably with diethyl phosphonoacetate, by After appropriate activation, in a suitable reaction solvent, preferably for the use of the active 1,1 – carbonyldiimidazole, 1,1 – carbonyldiimidazole – triazole or propane phosphonic acid anhydride, is preferred for the use of 1, 1 – carbonyl diimidazole. The solvent used may be, for example, tetrahydrofuran (THF), dimethylformamide (DMF) or ethyl acetate.
The amide may be connected through any possible approach for activation, i.e., for example, 1,1 _ carbonyldiimidazole, 1,1 – carbonyldiimidazole – triazole, DCC (N, N-dicyclohexyl carbodiimide ), EDC (N ‘_ (dimethylaminopropyl)-N-ethylcarbodiimide), TBTU (0 – (benzotriazol-1 – yl)-N, N, N’, N ‘ – pan tetramethyluronium tetrafluoroborate), thiazolidine-2 – thione, or through the use of thionyl chloride may be converted to the corresponding acyl chloride. If desired, activation may be used an organic base such as triethylamine or pyridine embodiment, and can additionally added DMAP (dimethylaminopyridine). Suitable solvents include DMF, THF, ethyl acetate, toluene, chlorinated hydrocarbons or mixtures thereof.

Example 1
[0078] {[4 - (3 - chloro-4 - fluoro - phenylamino) -7 - (⑶ - tetrahydrofuran _3_-yloxy) - quinazoline _6_ yl carbamoyl] methyl}-_ _ Diethyl
[0079]

A 3. 58kg of 1,1 _ carbonyldiimidazole (22.16 mol) was placed in 12.8 l of tetrahydrofuran, and at a temperature of 40 ° C was dissolved in it with 6.5 l of tetrahydrofuran, 4. 52kg (22. 16 mol) of diethyl phosphono acetic acid mixture. Temperature at 40 ° C the mixture was stirred for 30 minutes. The resulting solution was referred to as Solution A.
A 6. 39kg (17. 05 moles) of N4-(3_ _4_ chloro fluoro – phenyl) _7_ (tetrahydrofuran _3_ yloxy) quinazoline-4, 6 – diamine Add 26 5 of tetrahydrofuran at 40 ° C and the solution A were mixed and stirred at a temperature 30 ° C for 2 hours.To the suspension was added 64 l tert-butyl methyl ether and, after cooling to 20 ° C, the precipitate was removed by centrifugation. Using 16 liters of tetrahydrofuran and 16 l of a mixture of tert-butyl methyl ether, washed, and then washed with 32 liters of water and dried at 50 ° C.
[0082] Yield: 6. 58kg (69. 8%) of white crystals, the content = HPLC 99. IFl%
[0083] Example 2
[0084] (E) -4 – dimethylamino – D -2 – acid – [4 - (chloro-3_ _4_ fluoro - phenylamino) _7_ (⑶ - tetrahydrofuran-3 - yloxy) - quinoline yl-6 - yl] – amide
[0085]

[0086] A 5.6 l of 30% hydrochloric acid (53.17 mol) was added to 4.4 liters of water. Then the temperature is under 30 ° C was added dropwise over 20 minutes 4. 28kg 95% of (dimethylamino) _ acetaldehyde – diethyl acetal (26.59 mol).Temperature at 35 ° C the reaction solution was stirred for 8 hours was cooled to 5 ° C and kept under argon. This solution is called Solution B.
[0087] A 4. 55kg (68. 06 mol) of potassium hydroxide dissolved in 23.5 liters of water and cooled to _5 ° C. This solution is called Solution C.
[0088] A 5. 88kg (10. 63 mol) ((4_ (3_ _4_ chloro fluoro – phenylamino) _7_ (tetrahydrofuran _3_-yloxy) – quinazolin-6 – yl carbamoyl) – methyl)-phosphonic acid diethyl ester and 0.45kg _ lithium chloride (10.63 moles) was placed in 23.5 l of tetrahydrofuran and cooled to -7 ° C. Was added over 10 minutes a cold solution of C. Then _7 ° C temperature of the solution was added over 1 hour B. At _5 ° C temperature for 1 hour under stirring the reaction mixture was heated to 20 ° C and mixed with 15 liters of water. After cooling to; TC temperature, the suspension was suction filtered, the precipitate was washed with water and dried. Yield: 5.21kg The crude product, 100%, water content: 6.7%.
[0089] Using Titanium Dioxide / methyl cyclohexane embodiment the crystallization of the crude product.
[0090] Yield: 78%, purity: HPLC99. 4F1%, water content: 5.4%
[0091] Example 3
[0092] (E) -4 – dimethylamino – D -2 – acid – (4 – (chloro-3_ _4_ fluoro – phenylamino) ~ 7 ~ ((S) – tetrahydrofuran-3 – yl oxy) – quinazolin-6 – yl) – amide dimaleate
[0093] A 6. Okg (12. 35 mol) of (E_) _4_ _2_ dimethylamino acid _ D – (4_ (3_ _4_ chloro fluoro – phenylamino) -7 – ((S) – tetrahydrofuran-3 – yloxy) – quinazolin-6 – yl) – amide into 84 liters of ethanol and heated to 70 ° C, and dissolved in 36 l of ethanol and 2.94kg (25.31 moles) of maleic acid was mixed . At the beginning of crystallization, the first mixture was cooled to 20 ° C and stirred for 2 hours and then at 0 ° C temperature for 3 hours. Precipitate was suction filtered, washed with 19 l of ethanol at a temperature of 40 ° C in vacuo.
[0094] Yield: 8. Ilkg (91. 5%)
[0095] Melting point: 178 ° C
[0096] 1H-NMR (CD3OD): δ = 2. 47 + 2. 27 (m + m, 2H), 2. 96 (s, 6H), 4. 03 (m, 2Η), 4. 07 +3 . 92 (m + m, 2Η), 4. 18 +4. 03 (m + m, 2Η), 5. 32 (m, 1Η), 6. 26 (s, 4H), 6. 80 (m, 1H ), 6. 99 (m, 1H), 7 · 27 (s, 1Η), 7 · 30 (t, 1Η), 7 · 66 (m, 1Η), 7 · 96 (dd, 1Η), 8 · 62 (s, 1Η), 9 · 07 (s, 1Η) ppm
13
…………….
Examples:
Example 1
{[4 - (3-chloro-4-fluoro-phenylamino) -7 - ((S)-tetrahydrofuran-3-yloxy)-quinazolin-6-ylcarbamoyl]-methyl)-phosphonic acid diethyl ester

3.58 kg 1 ,1-carbonyldiimidazole (22.16 mole) were placed in 12.8 liters of tetrahydrofuran at 40 ° C with 4.52 kg (22.16 mol) diethylphosphonoacetic acid, dissolved in 6.5 liters of tetrahydrofuran, . The mixture is stirred for 30 minutes at 40 ° C. The solution thus obtained is referred to as solution A.
6.39 kg (17.05 mol) of N 4 - (3-chloro-4-fluoro-phenyl) -7 – (tetrahydrofuran-3-yloxy) quinazolin-4,6-diamine in 26.5 liters of tetrahydrofuran and submitted to 40 ° C and mixed with the solution A and stirred at 30 ° C for 2 hours. To 64 liters of suspension of tert -. Added butyl methyl ether and, after cooling to 20 ° C., the precipitate is removed by centrifugation. It is dried with a mixture of 16 liters and 16 liters of tetrahydrofuran tert-butyl methyl ether and then washed with 32 liters of water at 50 ° C. Yield: 6.58 kg (69.8%) of white crystals Assay: HPLC 99.1 area% Example 2
(E)-4-dimethylamino-but-2-enoic acid [4 – (3-chloro-4-fluoro-phenylamino) -7 – ((S) – tetrahvdrofuran-3-yloxy)-quinazolin-6yl1 amide

5.6 liters to 4.4 liters of water are added 30% hydrochloric acid (53.17 mol). Then 4.28 kg 95% pure (dimethylamino) acetaldehyde diethyl acetal (26.59 mol) at 30 ° C was added dropwise over 20 minutes. The reaction solution is stirred for 8 hours at 35 ° C, cooled to 5 ° C and kept under argon. This solution is referred to as solution B.
4.55 kg (68.06 mol) of potassium hydroxide are dissolved in 23.5 liters of water and cooled to -5 ° C. This solution is called solution C..
5.88 kg (10.63 mol) of ((4 – (3-chloro-4-fluoro-phenylamino) -7 – (tetrahydrofuran-3-yloxy) – quinazolin-6-ylcarbamoyl)-methyl)-phosphonic acid diethyl ester, and 0.45 kg lithium chloride (10.63 mole) were placed in 23.5 liters of tetrahydrofuran and cooled to -7 ° C. The cold solution C is added within 10 minutes. The solution B is added at -7 ° C over 1 hour. After stirring for one hour at -5 ° C, the reaction mixture is heated to 20 ° C and mixed with 15 liters of water. After cooling to 3 ° C, the suspension is filtered with suction, the precipitate washed with water and dried. Yield: 5.21 kg raw 100% Water content: 6.7%
The crystallization of the raw product is butyl acetate / methylcyclohexane yield: 78% HPLC purity 99.4 area%, water content 5.4% Example 3
(E)-4-dimethylamino-but-2-enoic acid (4 – (3-chloro-4-fluoro-pheny hvdrofuran-3-yloxy)-quinazolin-6YL) amide dimaleate
6.0 kg (12.35 mol) of (E)-4-dimethylamino-but-2-enoic acid (4 – (3-chloro-4-fluoro-phenyl-amino) -7 – ((S)-tetrahydrofuran- 3-yloxy) quinazolin-6YL)-amide are in 84 liters
Submitted ethanol and heated to 70 ° C and a solution of 2.94 kg (25.31 mol) of maleic acid in 36 liters of ethanol added.Following the onset of crystallization is first cooled to 20 ° C. and stirred for 2 hours, then 3 hours at 0 ° C. The precipitate is filtered off, washed with 19 liters of ethanol and dried in vacuum at 40 ° C.
Yield: 8.11 kg (91, 5%)
Mp: 178 ° C.
1 H NMR (CD 3 OD): δ = 2.47 + 2.27 (m + m, 2H), 2.96 (s, 6H), 4.03 (m, 2H), 4.07 + 3 , 92
(M + m, 2H), 4.18 + 4.03 (m + m, 2H), 5.32 (m, 1 H), 6.26 (s, 4H), 6.80 (m, 1 H ), 6.99 (m, 1 H), 7.27 (s, 1 H), 7.30 (t, 1 H), 7.66 (m, 1 H), 7.96 (dd, 1 H ), 8.62 (s, 1 H), 9.07 (s, 1H) ppm
…………..

U.S. Patent No. : 8,426,586 patent expires : October 10, 2029
WO200250043A1 (compound);
WO2003094921A2 (anticancer purposes);
WO2003066060A2 (anti-inflammatory purposes);
US2005085495A1 (process);
WO2005037824A2 (process);
WO2007085638A1 (process);
US2011207932A1 (process);
WO2011084796A2 (deuterated);
WO2012121764A1 (crystalline);
WO2013052157A1 (crystalline)
Chinese patents : CN1867564
CN101402631
5-30-2012
|
Amide derivative for inhibiting the growth of cancer cells
| |
6-15-2011
|
PROCESS FOR PREPARING AMINOCROTONYLAMINO-SUBSTITUTED QUINAZOLINE DERIVATIVES
| |
12-25-2009
|
METHOD FOR TREATING CANCER HARBORING EGFR MUTATIONS
| |
12-11-2009
|
QUINAZOLINE DERIVATIVES FOR THE TREATMENT OF CANCER DISEASES
| |
12-11-2009
|
COMBINATION TREATMENT OF CANCER COMPRISING EGFR/HER2 INHIBITORS
| |
9-12-2008
|
Multi-Functional Small Molecules as Anti-Proliferative Agents
| |
4-22-2005
|
Process for preparing amino crotonyl compounds
|
///////////////////////////////
4 SELUMETINIB

Selumetinib司美替尼
6-(4-bromo-2-chloroanilino)-7-fluoro-N-(2-hydroxyethoxy)-3-methylbenzimidazole-5-carboxamide
5-(4-Bromo-2-chlorophenylamino)-4-fluoro-1-methyl-1H-benzimidazole-6-carbohydroxamic acid 2-hydroxyethyl ester
6-(4-bromo-2-chloro- phenylamino)-7-fluoro-3 -methyl-3H-benzoimidazole-5-carboxylic acid (2-hydroxy- ethoxy)-amide
943332-08-9 (sulfate (1:1) salt) IS THE DRUG
Selumetinib, AZD6244, AZD-6244, ARRY-142886
Molecular Formula: C17H15BrClFN4O3 Molecular Weight: 457.681403
Non-small-cell lung cancer (NSCLC) is the most common type of lung cancer. In October, AstraZeneca began a phase III trial of selumetinib in patients with KRAS mutation-positive NSCLC. AstraZeneca has also partnered with Roche Molecular Systems to develop a device to detect these mutations.
Selumetinib (AZD6244) is a drug being investigated for the treatment of various types of cancer, for example non-small cell lung cancer (NSCLC).
The gene BRAF is part of the MAPK/ERK pathway, a chain of proteins in cells that communicates input from growth factors. Activating mutations in the BRAF gene, primarily V600E (meaning that the amino acid valine in position 600 is replaced by glutamic acid), are associated with lower survival rates in patients with papillary thyroid cancer. Another type of mutation that leads to undue activation of this pathway occurs in the gene KRAS and is found in NSCLC. A possibility of reducing the activity of the MAPK/ERK pathway is to block the enzyme MAPK kinase (MEK), immediately downstream of BRAF, with the drug selumetinib. More specifically, selumetinib blocks the subtypes MEK1 and MEK2 of this enzyme.[1]
Selumetinib is a novel, selective, non-ATP-competitive inhibitor of MEK1/2 currently in phase III clinical development at AstraZeneca for the oral treatment of non-small lung cancer with KRAS mutation. Additional phase II trials are under way at both AstraZeneca and Array BioPharma for the treatment of other oncological indications, including colorectal cancer, thyroid cancer and malignant melanoma. AstraZeneca is conducting phase I/II clinical trials for the treatment of Kaposi’s sarcoma (AIDS-related) in combination with highly active anti-retroviral therapy (HAART). Also, phase I trials are ongoing at the companies targeting several solid tumors, including skin, pancreatic, colon, lung and breast tumors. The National Cancer Institute (NCI) is also evaluating selumetinib for the treatment of thyroid cancer, ovary cancer, myeloid leukemia, glioma, multiple myeloma, metastatic uveal melanoma, sarcoma, pancreatic cancer, plexiform neurofibromas and for the treatment of recurrent or persistent endometrial cancer. Additional early clinical trials are under way at the Massachusetts General Hospital for the treatment of cancers with BRAF mutations. No recent development has been reported for phase II clinical trials for the treatment of metastatic pancreatic cancer.

In addition to thyroid cancer, BRAF-activating mutations are prevalent in melanoma (up to 59%), colorectal cancer (5–22%), serousovarian cancer (up to 30%), and several other tumor types.[2]
KRAS mutations appear in 20 to 30% of NSCLC cases and about 40% of colorectal cancer.[1]
. The National Cancer Institute (NCI) is also evaluating selumetinib for the treatment of thyroid cancer, ovary cancer, myeloid leukemia, glioma, multiple myeloma, metastatic uveal melanoma, sarcoma, pancreatic cancer, plexiform neurofibromas and for the treatment of recurrent or persistent endometrial cancer. Additional early clinical trials are under way at the Massachusetts General Hospital for the treatment of cancers with BRAF mutations. No recent development has been reported for phase II clinical trials for the treatment of metastatic pancreatic cancer.
A Phase II clinical trial about selumetinib in NSCLC has been completed in September 2011;[3] one about cancers with BRAF mutations is ongoing as of June 2012.[4]
Selumetinib appears to efficiently target cancers with overactivation of MEK and associated cell signaling pathways. According to laboratory studies, selumetinib has an effect on human tumors at nanomolar concentrations. Potential advantages of selumetinib over marketed therapies include improved efficacy linked to a novel mechanism and ease of use based on the drug candidate’s oral formulation.
In 2013, AstraZeneca acquired exclusive worldwide rights to selumetinib from Array BioPharma.
In 2013, AstraZeneca acquired exclusive worldwide rights to selumetinib from Array BioPharma.
AZD6244 (Selumetinib)
6-(4-Bromo-2- chloro-ρhenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid (2-hydroxy- ethoxy)-amide, or “Compound 1″, is exemplified in WO 03/077914 and possesses the following structural formula:


…………………………..
Example 10

6-(4-Bromo-2-chloro-phenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid (2-hydroxy-ethoxy)-amide (29c)
Step A. 6-(4-Bromo-2-chloro-phenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid methyl ester 9a and 6-(4-Bromo-2-chloro-phenylamino)-7-fluoro-1-methyl-1H-benzoimidazole-5-carboxylic acid methyl ester
A solution of 6-(4-bromo-2-chloro-phenylamino)-7-fluoro-3H-benzoimidazole-5-carboxylic acid methyl ester 8b (150 mg, 0.38 mmol), iodomethane (28 μL, 0.45 mmol) and potassium carbonate (78 mg, 0.56 mmol) in dimethylformamide (1.5 mL) is stirred at 75° C. for one hour. The reaction mixture is diluted with ethyl acetate, washed with saturated aqueous potassium carbonate (2×), brine, and dried (Na2SO4). Flash column chromatography (20:1 methylene chloride/ethyl acetate) provides 56 mg (36%) of the more mobile 6-(4-bromo-2-chloro-phenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid methyl ester 9a as a white solid. 19F NMR (376 MHz, CD3OD)-133.5 (s). MS APCI (+) m/z 412, 414 (M+, Br pattern) detected. Also isolated is 54 mg (35%) of 6-(4-bromo-2-chloro-phenylamino)-7-fluoro-1-methyl-1H-benzoimidazole-5-carboxylic acid methyl ester as a white solid. 19F NMR (376 MHz, CD3OD)-139.9 (s). MS APCI (+) m/z 412, 414 (M+, Br pattern) detected.
Step B. 6-(4-Bromo-2-chloro-phenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid 10c
6-(4-Bromo-2-chloro-phenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid methyl ester 9a (56 mg, 0.14 mmol) is dissolved into 2:1 THF/water (3 mL) and NaOH (0.55 mL, 1.0 M aqueous solution, 0.55 mmol) is added. After stirring for two hours the reaction is reduced to one quarter initial volume via rotary evaporation and the remainder diluted to 50 mL with water. The aqueous solution is acidified to pH 2 by the addition of 1.0 M aqueous HCl and extracted with 1:1 tetrahydrofuran/ethyl acetate (3×), dried (Na2SO4) and concentrated under reduced pressure to provide 43 mg (79%) pure carboxylic acid as an off white solid. MS ESI (+) m/z 397, 398 (M+, Br pattern) detected.
Step C: 6-(4-Bromo-2-chloro-phenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid (2-vinyloxy-ethoxy)-amide 29a
6-(4-Bromo-2-chloro-phenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid 10c (2.00 g, 5.0 mmol), O-(2-vinyloxy-ethyl)-hydroxylamine (0.776 g, 7.5 mmol), HOBt (0.88 g, 6.5 mmol), triethylamine (1.61 mL, 2.3 mmol) and EDCI (1.3 g, 6.5 mmol) are dissolved in dimethylformamide (52 mL) and stirred at room temperature for 48 hours. The reaction mixture is diluted with ethyl acetate, washed with water (3×), saturated potassium carbonate (2×), saturated ammonium chloride (2×), brine, dried (Na2SO4) and concentrated under reduced pressure to an off-white solid. Trituration of the solid with diethyl ether provides 2.18 g (90%) desired product as an off-white solid. MS ESI (+) m/z 483, 485 (M+ Br pattern) detected.
Step D: 6-(4-Bromo-2-chloro-phenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid (2-hydroxy-ethoxy)-amide 29c
Hydrochloric acid (14 mL, 1.0 M aqueous solution, 14 mmol) is added to a suspension of 6-(4-bromo-2-chloro-phenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid (2-vinyloxy-ethoxy)-amide 29a (2.18 g, 4.50 mmol) in ethanol (50 mL) and the reaction mixture allowed to stir for 24 hours. The reaction mixture is concentrated to dryness by rotary evaporation and the solids partitioned between 3:1 ethyl acetate/tetrahydrofuran and saturated potassium carbonate. The aqueous phase is extracted with 3:1 ethyl acetate/tetrahydrofuran (3×), the combined organics dried (Na2SO4), and concentrated to provide 2.11 g (100%) 6-(4-bromo-2-chloro-phenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid (2-hydroxy-ethoxy)-amide as an off-white solid. MS ESI (+) m/z 457, 459 (M+, Br pattern) detected. 1H NMR (400 MHz, MeOH-d4) δ8.26 (s, 1H), 7.78 (s, 1H), 7.57 (d, 1H), 7.24 (dd, 1H), 6.40 (dd, 1H), 3.86 (s, 3H), 3.79 (m, 2H), 3.49 (m, 2H). 19F NMR (376 MHz, MeOH-d4)-133.68 (s).
…………
Scheme 1


Scheme la

Scheme 2

Scheme 3

17 18
Scheme 4

25
Scheme 5


Example 1 and in this Example 9 by using the appropriate carboxylic acid and the appropriate hydroxylamine:

Example 10

6-(4-Bromo-2-chloro-phenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid (2-hydroxy-ethoxy)-amide (29c)Step A. 6-(4-Bromo-2-chloro-phenylamino)- 7-fluoro-3-methyl-3H-benzoimidazole-5- carboxylic acid methyl ester 9a and 6-(4-Bromo-2-chloro-phenylamino)-7-fluoro-l- methyl-lH-benzoimidazole-5-carboxylic acid methyl ester
A solution of 6-(4-bromo-2-chloro-phenylamino)-7-fluoro-3H-benzoimidazole-5-
carboxylic acid methyl ester 8b (150 mg, 0.38 mmol), iodomethane (28 μL, 0.45 mmol)
and potassium carbonate (78 mg, 0.56 mmol) in dimethylformamide (1.5 mL) is stirred at
75 °C for one hour. The reaction mixture is diluted with ethyl acetate, washed with saturated aqueous potassium carbonate (2x), brine, and dried (Na SO ). Flash column chromatography (20:1 methylene chloride/ethyl acetate) provides 56 mg (36%) of the
more mobile 6-(4-bromo-2-chloro-phenylamino)-7-fluoro-3 -methyl-3H-benzoimidazole-
5-carboxylic acid methyl ester 9a as a white solid. 19F NMR (376 MHz, CD3OD) -133.5
(s). MS APCI (+) m/z 412, 414 (M+, Br pattern) detected. Also isolated is 54 mg (35%)
of 6-(4-bromo-2-chloro-phenylamino)-7-fluoro-l-methyl-lH-benzoimidazole-5- carboxylic acid methyl ester as a white solid. 19F NMR (376 MHz, CD3OD) -139.9 (s).
MS APCI (+) m/z 412, 414 (M+, Br pattern) detected.
Step B. 6-(4-Bromo-2-chloro-phenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5- carboxylic acid 10c
6-(4-Bromo-2-chloro-phenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5- carboxylic acid methyl ester 9a (56 mg, 0.14 mmol) is dissolved into 2:1 THF/water (3 mL ) and NaOH (0.55 mL, 1.0 M aqueous solution, 0.55 mmol) is added. After stirring for two hours the reaction is reduced to one quarter initial volume via rotary evaporation and the remainder diluted to 50 mL with water. The aqueous solution is acidified to pH 2 by the addition of 1.0 M aqueous HCl and extracted with 1 : 1 tetrahydrofuran/ethyl acetate (3x), dried (Na2SO4) and concentrated under reduced pressure to provide 43 mg (79%) pure carboxylic acid as an off white solid. MS ESI (+) m/z 397, 398 (M+, Br pattern) detected.
Step C: 6-(4-Bromo-2-chloro-phenylamino)~ 7-fluoro-3-methyl-3H-benzoimidazole-5- carboxylic acid (2-vinyloxy-ethoxy)-amide 29a
6-(4-Bromo-2-chloro-phenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5- carboxylic acid 10c (2.00 g, 5.0 mmol), O-(2-vinyloxy-ethyl)-hydroxylamine (0.776 g, 7.5 mmol), HOBt (0.88 g, 6.5 mmol), triethylamine (1.61 mL, 2.3 mmol) and EDCI (1.3 g, 6.5 mmol) are dissolved in dimethylformamide (52 mL) and stirred at room temperature for 48 hours. The reaction mixture is diluted with ethyl acetate, washed with water (3x), saturated potassium carbonate (2x), saturated ammonium chloride (2x), brine, dried (Na2SO4) and concentrated under reduced pressure to an off-white solid. Trituration of the solid with diethyl ether provides 2.18 g (90%) desired product as an off- white solid. MS ESI (+) m/z 483, 485 (M+ Br pattern) detected.
Step D: 6-(4-Bromo-2-chloro-phenylamino)- 7-fluoro-3-methyl-3H-benzoimidazole-5- carboxylic acid (2-hydroxy-ethoxy) -amide 29c
Hydrochloric acid (14 mL, 1.0 M aqueous solution, 14 mmol) is added to a suspension of 6-(4-bromo-2-chloro-phenylamino)-7-fluoro-3 -methyl-3H-benzoimidazole -5-carboxylic acid (2-vinyloxy-ethoxy)-amide 29a (2.18 g, 4.50 mmol) in ethanol (50 mL) and the reaction mixture allowed to stir for 24 hours. The reaction mixture is concentrated to dryness by rotary evaporation and the solids partitioned between 3:1 ethyl acetate/tefrahydrofuran and saturated potassium carbonate. The aqueous phase is extracted with 3:1 ethyl acetate/tefrahydrofuran (3x), the combined organics dried (Na SO4), and concentrated to provide 2.11 g (100%) 6-(4-bromo-2-chloro- phenylamino)-7-fluoro-3 -methyl-3H-benzoimidazole-5-carboxylic acid (2-hydroxy- ethoxy)-amide as an off-white solid. MS ESI (+) m/z 457, 459 (M+, Br pattern) detected. 1H NMR (400 MHz, MeOH-c^) δ 8.26 (s, IH), 7.78 (s, IH), 7.57 (d, IH), 7.24 (dd, IH), 6.40 (dd, IH), 3.86 (s, 3H), 3.79 (m, 2H), 3.49 (m, 2H). 19F NMR (376 MHz, MeOH-d4) -133.68 (s).
………………
Example 1
Preparation of the Hydrogen sulfate salt of Compound 1

[0076] To a stirred suspension of 6-(4-bromo-2-chloro-phenylamino)-7-fiuoro-3- methyl-3H-benzoimidazole-5-carboxylic acid (2-hydroxy-ethoxy)-amide (100 g, 0.206 mol) (obtainable as described in Example 10 of WO 03/077914, which is incorporated herein by reference and as described below) in 2-butanone (680 mL) and water (115 mL) at 0-5 0C was added sulfuric acid (12.3 mL, 0.226 mol) followed by water (5 mL) maintaining a temperature of 10 °C or lower. The stirred mixture was heated to 65 0C and held for 30 minutes before filtering to remove any extraneous matter. The filter was washed with a mixture of 2-butanone (85 mL) and water (15 mL). The combined filtrates were heated to 72 0C before adding 2-butanone (500 mL) maintaining a temperature of between 60-72 0C. The resulting mixture was distilled at atmospheric pressure (approximate distillation temperature 73-74°C) until 500 mL of distillate had been collected.
[0077] A second aliquot of 2-butanone (500 mL) was added, maintaining the temperature of the mixture above 70 0C. The resulting mixture was distilled again until 250 mL of distillate had collected. The mixture was cooled to 0-5 0C over approximately 1 hour. The resulting slurry was filtered, washed with 2-butanone (240 mL) and dried under reduced pressure at 50 0C, until a constant weight was achieved, to give 6-(4-bromo-2-chloro- phenylamino)-7-fiuoro-3-methyl-3H-benzoimidazole-5-carboxylic acid (2-hydroxy-ethoxy)- amide hydrogen sulfate (103.5 g, 0.186 mol, 90% yield) as an off white crystalline solid.1H NMR (400 MHz, D6 DMSO) δ 3.58 (2H, t, CH2OH), 3.89 (2H, t, CH2ON), 3.99 (3H, s, CH3), 6.47 (IH, dd, ArH), 7.29 (IH, dd, ArH), 7.63 (IH, d, ArH), 7.91 (IH, s, ArH), 7.96 (3H, br, ROH, NH, SOH), 8.10 (IH, br, ArNH), 8.94 (IH, s, NCHN), 11.79 (IH, s, ONH). 13C NMR (100 MHz, D6 DMSO) δ 32.1 (CH3), 58.5 (CH2OH), 77.3 (CH2ON), 108.2 (CH), 109.6 (CBr), 115.8 (CH), 120.6 (CCl), 122.0 (C), 125.0 (CC=O), 129.4 (C), 130.5 (CH), 131.1 (CH), 132.3 (C), 140.6 (C), 145.8 (CF), 146.5 (CH), 164.2 (C=O). [0078] The results of the infrared analysis are shown in Figure 2. Spectral assignments axe summarized in Table 1.
Table 1
Wavenumber (cm“ ) Assignment 3,255 Includes the O-H stretching vibration of the primary alcohol group and the N-H stretching vibrations of the secondary aromatic amine and secondary amide groups.
3,200 – 2,700 Includes =C-H stretching vibrations of the aromatic ring and benzimidazole group and the aliphatic C-H stretching vibrations.
2,700 – 2,300 Includes the multiple NH+ stretching vibrations of the benzimidazole 1 : 1 sulfate salt group.
1,673 C=O stretching vibrations of the secondary amide group where
1,653 the carbonyl group is subject to different environmental effects such as hydrogen bonding.
1,640 – 1,370 Includes the C=C aromatic ring stretching vibrations, the C=C and C=N stretching vibrations of the benzimidazole group, the
O-H deformation vibration of the primary alcohol group and the aliphatic C-H deformation vibrations.
1,570 The CNH combination band of the secondary amide group.
1,506 Includes the CNH bending vibration of the secondary aromatic amine group.
1 ,213 The aryl C-F stretching vibration.
1,189 The asymmetric SO3 “ stretching vibration of the benzimidazole
1 : 1 sulfate salt group. 1,100 – 1,000 Includes the C-O stretching vibration of the primary alcohol group and the aryl C-Br stretching vibration. 1,011 The symmetric SO3 “ stretching vibration of the benzimidazole
1 :1 sulfate salt group. 920 – 600 Includes the C-H wag vibrations and C=C ring bending vibrations of the 1,2,4-trisubtituted aromatic ring and the benzimidazole group. 888 Includes the S-O(H) stretching vibration of the benzimidazole
1 : 1 sulfate salt group. Example IA
Preparation of the Hydrogen sulphate salt of Compound 1
[0079] Sulfuric acid (1.52 ml, 27.86 mmol) was added to a stirred suspension of 6-(4- bromo-2-chlorophenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid (2- hydroxyethoxy)-amide (1O g, 0.0214 mol) (obtainable as described in Example 10 of WO 03/077914, which is incorporated herein by reference and as described below) in tetrahydrofuran (THF) (62 ml) and water (8 ml) whilst maintaining a temperature of 10 0C or lower. The stirred mixture was heated to 65 0C and held for 30 minutes before filtering to remove any extraneous matter. THF (150 ml) was then added to the mixture maintaining the temperature above 60 0C. The mixture was then cooled to 0-5 0C over approximately 2 hour. The resulting slurry was filtered, washed with THF (30 ml) and dried under reduced pressure at 50 0C until a constant weight was achieved, to give 6-(4-bromo-2-chlorophenylamino)-7- fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid (2-hydroxyethoxy)-amide hydrogen sulfate (9.81g, 0.17 mol, 82% yield) as an off white crystalline solid. The material was the same as that produced in Example 1 above.
References
- Troiani, T.; Vecchione, L.; Martinelli, E.; Capasso, A.; Costantino, S.; Ciuffreda, L. P.; Morgillo, F.; Vitagliano, D.; d’Aiuto, E.; De Palma, R.; Tejpar, S.; Van Cutsem, E.; De Lorenzi, M.; Caraglia, M.; Berrino, L.; Ciardiello, F. (2012). “Intrinsic resistance to selumetinib, a selective inhibitor of MEK1/2, by cAMP-dependent protein kinase a activation in human lung and colorectal cancer cells”. British Journal of Cancer 106 (10): 1648–1659.doi:10.1038/bjc.2012.129. PMC 3349172. PMID 22569000.
- Davies, H.; Bignell, G. R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M. J.; Bottomley, W.; Davis, N.; Dicks, E.; Ewing, R.; Floyd, Y.; Gray, K.; Hall, S.; Hawes, R.; Hughes, J.; Kosmidou, V.; Menzies, A.; Mould, C.; Parker, A.; Stevens, C.; Watt, S.; Hooper, S.; Wilson, R.; Jayatilake, H.; Gusterson, B. A.; Cooper, C.; Shipley, J. (2002). “Mutations of the BRAF gene in human cancer”. Nature 417 (6892): 949–954. doi:10.1038/nature00766. PMID 12068308.
- Jump up^ ClinicalTrials.gov NCT00890825 Comparison of AZD6244 in Combination With Docetaxel Versus Docetaxel Alone in KRAS Mutation Positive Non Small Cell Lung Cancer (NSCLC) Patients
- Jump up^ ClinicalTrials.gov NCT00888134 AZD6244 in Cancers With BRAF Mutations
- Journal of the American Chemical Society, 2013 , vol. 135, 35 p. 12994 – 12997
- 8-1-2013Identification of potent Yes1 kinase inhibitors using a library screening approach.Bioorganic & medicinal chemistry letters
| WEDGE S R ET AL: “AZD2171: A HIGHLY POTENT, ORALLY BIOAVAILABLE, VASCULAR ENDOTHELIAL GROWTH FACTOR RECEPTOR-2 TYROSINE KINASE INHIBITOR FOR THE TREATMENT OF CANCER“, CANCER RESEARCH, AMERICAN ASSOCIATION FOR CANCER RESEARCH, US, vol. 65, no. 10, 15 May 2005 (2005-05-15), pages 4389-4400, XP008066714, ISSN: 0008-5472, DOI: 10.1158/0008-5472.CAN-04-4409 | ||
| 52 | * | WEDGE STEPHEN R ET AL: “ZD6474 inhibits vascular endothelial growth factor signaling, angiogenesis, and tumor growth following oral administration“, CANCER RESEARCH, AMERICAN ASSOCIATION FOR CANCER RESEARCH, US, vol. 62, no. 16, 15 August 2002 (2002-08-15), pages 4645-4655, XP002425560, ISSN: 0008-5472 |
| 53 | WEDGE, S.R. ET AL.: ‘ZD6474 inhibits vascular endothelial growth factor signaling, angiogenesis, and tumor growth following oral administration‘ CANCER RES vol. 62, 2002, pages 4645 – 4655 |
- Ho, Alan L.; Grewal, Ravinder K.; Leboeuf, Rebecca; Sherman, Eric J.; Pfister, David G.; Deandreis, Desiree; Pentlow, Keith S.; Zanzonico, Pat B. et al. (2013). “Selumetinib-Enhanced Radioiodine Uptake in Advanced Thyroid Cancer”. New England Journal of Medicine 368 (7): 623–32. doi:10.1056/NEJMoa1209288. PMC 3615415.PMID 23406027.
1-30-2009
|
TOSYLATE SALT OF 6- (4-BR0M0-2-CHL0R0PHENYLAMIN0) -7-FLUORO-N- (2-HYDROXYETHOXY) -3-METHYL-3H-BENZIMI DAZOLE- 5 – CARBOXAMIDE , MEK INHIBITOR USEFUL IN THE TREATMENT OF CANCER
| |
9-17-2008
|
N3 alkylated benzimidazole derivatives as MEk inhibitors
| |
6-27-2007
|
N3 alkylated benzimidazole derivatives as MEK inhibitors
| |
12-19-2003
|
N3 alkylated benzimidazole derivatives as MEK inhibitors
|
6-6-2012
|
METHOD OF TREATMENT USING N3 ALKYLATED BENZIMIDAZOLE DERIVATIVES AS MEK INHIBITORS
| |
6-6-2012
|
COMPOSITIONS COMPRISING N3 ALKYLATED BENZIMIDAZOLE DERIVATIVES AS MEK INHIBITORS AND METHODS OF USE THEREOF
| |
5-16-2012
|
N3 ALKYLATED BENZIMIDAZOLE DERIVATIVES AS MEK INHIBITORS
| |
8-24-2011
|
N3 ALKYLATED BENZIMIDAZOLE DERIVATIVES AS MEK INHIBITORS
| |
7-6-2011
|
N3 ALKYLATED BENZIMIDAZOLE DERIVATIVES AS MEK INHIBITORS
| |
11-31-2010
|
N3 ALKYLATED BENZIMIDAZOLE DERIVATIVES AS MEK INHIBITORS
| |
8-18-2010
|
N3 ALKYLATED BENZIMIDAZOLE DERIVATIVES AS MEK INHIBITORS
| |
5-28-2010
|
COMBINATION THERAPY COMPRISING AZD2171 AND AZD6244 OR MEK-INHIBITOR II
| |
10-2-2009
|
PHARMACEUTICAL COMPOSITION 271
| |
8-19-2009
|
N3 ALKYLATED BENZIMIDAZOLE DERIVATIVES AS MEK INHIBITORS
|
5 ALECTINIB

Alectinib (AF802, CH5424802, RG7853, RO5424802)
CAS 1256580-46-7 FREE
1256589-74-8 (Alectinib Hydrochloride)
9-Ethyl-6,11-dihydro-6,6-dimethyl-8-[4-(4-morpholinyl)-1-piperidinyl]-11-oxo-5H-benzo[b]carbazole-3-carbonitrile
| Formula: | C30H34N4O2 |
| M.Wt: | 482.62 |
Mechanism of Action:ALK inhibitor
Indication:Non-small cell lung cancer (NSCLC)
Current Status:Phase II (US,EU,UK), NDA(Japan)
Company:中外製薬株式会社 (Chugai), Roche
Indication:Non-small cell lung cancer (NSCLC)
Current Status:Phase II (US,EU,UK), NDA(Japan)
Company:中外製薬株式会社 (Chugai), Roche
Japan First to Approve Alectinib for ALK+ NSCLC
Roche announced that the Japanese Ministry of Health, Labor and Welfare (MHLW) has approved alectinib for the treatment of people living with non-small cell lung cancer (NSCLC) that is anaplastic lymphoma kinase fusion gene-positive (ALK+). The approval was based on results from a Japanese Phase 1/2 clinical study (AF-001JP) for people whose tumors were advanced, recurrent or could not be removed completely through surgery (unresectable).

| Company | Chugai Pharmaceutical Co. Ltd. |
| Description | Anaplastic lymphoma kinase (ALK) inhibitor |
| Molecular Target | Anaplastic lymphoma kinase (ALK) |
| Mechanism of Action | Anaplastic lymphoma kinase (Ki-1) (ALK) inhibitor |
| Therapeutic Modality | Small molecule |
| Latest Stage of Development | Registration |
| Standard Indication | Non-small cell lung cancer (NSCLC) |
| Indication Details | Treat advanced ALK-positive non-small cell lung cancer (NSCLC); Treat non-small cell lung cancer (NSCLC); Treat unresectable progressive or recurrent ALK-positive non-small cell lung cancer (NSCLC) |
| Regulatory Designation |
U.S. - Breakthrough Therapy (Treat advanced ALK-positive non-small cell lung cancer (NSCLC));
Japan - Orphan Drug (Treat advanced ALK-positive non-small cell lung cancer (NSCLC)); Japan - Orphan Drug (Treat unresectable progressive or recurrent ALK-positive non-small cell lung cancer (NSCLC)); Japan - Standard Review (Treat advanced ALK-positive non-small cell lung cancer (NSCLC)) |
| Partner |

Alectinib (also known as CH5424802,RO5424802), a second generation oral inhibitor of anaplastic lymphoma kinase (ALK), is being developed by Chugai and Roche for the treatment of patients with ALK-positive non-small cell lung cancer (NSCLC) that has progressed on Xalkori (Crizotinib).
Alectinib was discovered by Chugai Pharmaceutical Co. Ltd. Chugai became a subsidiary of Roche in 2002 and the Swiss group currently owns 59.9 percent of the company.
On October 8, 2013, Chugai Pharmaceutical announced that it has filed a new drug application to Japan’s Ministry of Health, Labour and Welfare (MHLW) for alectinib hydrochloride for the treatment of ALK fusion gene positive non-small cell lung cancer (NSCLC).
IT is a potent and selective ALK inhibitor with IC50 of 1.9 nM.Alterations in the anaplastic lymphoma kinase (ALK) gene have been implicated in human cancers. Among these findings, the fusion gene comprising EML4 and ALK has been identified in non-small cell lung cancer (NSCLC) and fusion of ALK to NPM1 has been observed in anaplastic large cell lymphoma (ALCL). The possibility of targeting ALK in human cancer was advanced with the launch of crizotinib for NSCLC in the U.S. in 2011. The development of resistance to crizotinib in tumors, however, has led to the need for second-generation ALK inhibitors. One of these, alectinib hydrochloride, has been found to be an orally active, potent and highly selective ALK inhibitor with activity in ALK-driven tumor models. Alectinib has shown preclinical activity against cancers with ALK gene alterations, including NSCLC cells expressing the EML4-ALK fusion and ALCL cells expressing the NPM-ALK fusion. Alectinib was well tolerated and active in a phase I/II study conducted in Japan in patients with ALK-rearranged advanced NSCLC and in patients with ALK-positive NSCLC who had progressed on crizotinib. Alectinib has been submitted for approval in Japan for the treatment of ALK fusion gene-positive NSCLC and is in phase I/II development for ALK-rearranged NSCLC in the U.S.

.................

......................
WO2012023597
(Preparation 30)
Compound F6-20
9 - ethyl-6, 6 - dimethyl-8 - (4 - morpholin-4 - yl - piperidin-1 - yl) -11 - oxo-6 ,11 - dihydro-5H-benzo [b] carbazol-3 - carbonitrile
Compound F6-20
9 - ethyl-6, 6 - dimethyl-8 - (4 - morpholin-4 - yl - piperidin-1 - yl) -11 - oxo-6 ,11 - dihydro-5H-benzo [b] carbazol-3 - carbonitrile

Under the same conditions as the synthesis of the compound B3-13-1, and the title compound was synthesized from compound F5-49.
1 H-NMR (400MHz, DMSO-D 6) δ: 12.70 (1H, s), 8.32 (1H, d, J = 7.9 Hz), 8.04 (1H, s), 8.00 (1H, s), 7.61 (1H , d, J = 8.5 Hz), 7.34 (1H, s), 3.64-3.57 (4H, m), 3.27-3.18 (2H, m), 2.82-2.66 (4H, m), 2.39-2.28 (1H, m ), 1.96-1.87 (2H, m), 1.76 (6H, s), 1.69-1.53 (2H, m), 1.29 (3H, t, J = 7.3 Hz)
LCMS: m / z 483 [M + H] +
HPLC retention time: 1.98 minutes (analysis conditions U)
1 H-NMR (400MHz, DMSO-D 6) δ: 12.70 (1H, s), 8.32 (1H, d, J = 7.9 Hz), 8.04 (1H, s), 8.00 (1H, s), 7.61 (1H , d, J = 8.5 Hz), 7.34 (1H, s), 3.64-3.57 (4H, m), 3.27-3.18 (2H, m), 2.82-2.66 (4H, m), 2.39-2.28 (1H, m ), 1.96-1.87 (2H, m), 1.76 (6H, s), 1.69-1.53 (2H, m), 1.29 (3H, t, J = 7.3 Hz)
LCMS: m / z 483 [M + H] +
HPLC retention time: 1.98 minutes (analysis conditions U)
Hydrochloride 9 of compound F6-20 - ethyl-6, 6 - dimethyl-8 - (4 - morpholin-4 - yl - piperidin-1 - yl) -11 - oxo-6 ,11 - dihydro-5H-benzo [b I was dissolved at 60 ℃ in a mixture of 10 volumes of methyl ethyl ketone, 3 volumes of water and acetic acid volume 4-carbonitrile -] carbazol-3. I was dropped hydrochloric acid (2N) 1 volume of solution. After stirring for 30 minutes at 60 ℃, and the precipitated solid was filtered and added dropwise to 25 volume ethanol, 9 - Dry ethyl -6,6 - dimethyl-8 - (4 - morpholin-4 - yl - piperidin-1 - yl) I got a one-carbonitrile hydrochloride - 11 - oxo-6 ,11 - dihydro-5H-benzo [b] carbazol-3. Ethyl-6, 6 - 9 - obtained dimethyl-8 - (4 - morpholin-4 - yl - piperidin-1 - yl) -11 - oxo-6 ,11 - dihydro-5H-benzo [b] carbazol-3 - I was pulverized with a jet mill carbonitrile monohydrochloride.
1 H-NMR (400MHz, DMSO-D 6) δ: 12.78 (1H, s), 10.57 (1H, br.s), 8.30 (1H, J = 8.4 Hz), 8.05 (1H, s), 7.99 (1H , s), 7.59 (1H, d, J = 7.9 Hz), 7.36 (1H, s) ,4.02-3 .99 (2H, m) ,3.84-3 .78 (2H, m) ,3.51-3 .48 (2H, m), 3.15-3.13 (1H, s) ,2.83-2 .73 (2H, s) ,2.71-2 .67 (2H, s) ,2.23-2 .20 (2H, m) ,1.94-1 .83 (2H, m), 1.75 (6H, s ), 1.27 (3H, t, J = 7.5 Hz)
FABMS: m / z 483 [M + H] +
1 H-NMR (400MHz, DMSO-D 6) δ: 12.78 (1H, s), 10.57 (1H, br.s), 8.30 (1H, J = 8.4 Hz), 8.05 (1H, s), 7.99 (1H , s), 7.59 (1H, d, J = 7.9 Hz), 7.36 (1H, s) ,4.02-3 .99 (2H, m) ,3.84-3 .78 (2H, m) ,3.51-3 .48 (2H, m), 3.15-3.13 (1H, s) ,2.83-2 .73 (2H, s) ,2.71-2 .67 (2H, s) ,2.23-2 .20 (2H, m) ,1.94-1 .83 (2H, m), 1.75 (6H, s ), 1.27 (3H, t, J = 7.5 Hz)
FABMS: m / z 483 [M + H] +
I was dissolved at 90 ℃ to 33 volume dimethylacetamide F6-20 F6-20 mesylate. Was added to 168 volumes mesylate solution (2 N) 1.2 volume, ethyl acetate solution was stirred for 4 hours. The filtered crystals were precipitated, and dried to obtain a F6-20 one mesylate. I was milled in a jet mill F6-20 one mesylate salt was obtained.
........................
Journal of Medicinal Chemistry, 54(18), 6286-6294; 2011
| WO2002043704A1 * | 30 Nov 2001 | 6 Jun 2002 | Yasuki Kato | Composition improved in solubility or oral absorbability |
| WO2008051547A1 * | 23 Oct 2007 | 2 May 2008 | Cephalon Inc | Fused bicyclic derivatives of 2,4-diaminopyrimidine as alk and c-met inhibitors |
| WO2009073620A2 * | 1 Dec 2008 | 11 Jun 2009 | Newlink Genetics | Ido inhibitors |
| WO2010143664A1 * | 9 Jun 2010 | 16 Dec 2010 | Chugai Seiyaku Kabushiki Kaisha | Tetracyclic compound |
| JP2008280352A | Title not available | |||
| JP2009100783A | Title not available | |||
| JPH0892090A * | Title not available |
References
|
1: Ignatius Ou SH, Azada M, Hsiang DJ, Herman JM, Kain TS, Siwak-Tapp C, Casey C, He J, Ali SM, Klempner SJ, Miller VA. Next-generation sequencing reveals a Novel NSCLC ALK F1174V mutation and confirms ALK G1202R mutation confers high-level resistance to alectinib (CH5424802/RO5424802) in ALK-rearranged NSCLC patients who progressed on crizotinib. J Thorac Oncol. 2014 Apr;9(4):549-53. doi: 10.1097/JTO.0000000000000094. PubMed PMID: 24736079.
2: Gouji T, Takashi S, Mitsuhiro T, Yukito I. Crizotinib can overcome acquired resistance to CH5424802: is amplification of the MET gene a key factor? J Thorac Oncol. 2014 Mar;9(3):e27-8. doi: 10.1097/JTO.0000000000000113. PubMed PMID: 24518097.
3: Latif M, Saeed A, Kim SH. Journey of the ALK-inhibitor CH5424802 to phase II clinical trial. Arch Pharm Res. 2013 Sep;36(9):1051-4. doi: 10.1007/s12272-013-0157-8. Epub 2013 May 23. Review. PubMed PMID: 23700294.
4: Seto T, Kiura K, Nishio M, Nakagawa K, Maemondo M, Inoue A, Hida T, Yamamoto N, Yoshioka H, Harada M, Ohe Y, Nogami N, Takeuchi K, Shimada T, Tanaka T, Tamura T. CH5424802 (RO5424802) for patients with ALK-rearranged advanced non-small-cell lung cancer (AF-001JP study): a single-arm, open-label, phase 1-2 study. Lancet Oncol. 2013 Jun;14(7):590-8. doi: 10.1016/S1470-2045(13)70142-6. Epub 2013 Apr 30. PubMed PMID: 23639470.
5: Kinoshita K, Asoh K, Furuichi N, Ito T, Kawada H, Hara S, Ohwada J, Miyagi T, Kobayashi T, Takanashi K, Tsukaguchi T, Sakamoto H, Tsukuda T, Oikawa N. Design and synthesis of a highly selective, orally active and potent anaplastic lymphoma kinase inhibitor (CH5424802). Bioorg Med Chem. 2012 Feb 1;20(3):1271-80. doi: 10.1016/j.bmc.2011.12.021. Epub 2011 Dec 22. PubMed PMID: 22225917.
6: Sakamoto H, Tsukaguchi T, Hiroshima S, Kodama T, Kobayashi T, Fukami TA, Oikawa N, Tsukuda T, Ishii N, Aoki Y. CH5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutant. Cancer Cell. 2011 May 17;19(5):679-90. doi: 10.1016/j.ccr.2011.04.004. PubMed PMID: 21575866.
Gadgeel S, Ou SH, Chiappori A, et al: A phase I dose escalation study of a new ALK inhibitor, CH542480202, in ALK+ non-small cell lung cancer patients who have failed crizotinib. Abstract O16.06. Presented at the 15th World Conference on Lung Cancer, Sydney, Australia, October 29, 2013.
Ou SH, Gadgeel S, Chiappori AA, et al: Consistent therapeutic efficacy of CH5424802/RO5424802 in brain metastases among crizotinib-refractory ALK-positive non-small cell lung cancer patients in an ongoing phase I/II study. Abstract O16.07. Presented at the 15th World Conference on Lung Cancer, Sydney, Australia, October 29, 2013.
Kinoshita, Kazuhiro et al,Preparation of tetracyclic compounds such as 11-oxo-5,6-dihydrobenzo[b]carbazole-3-carbonitrile derivatives as anaplastic lymphoma kinase (ALK) inhibitors,Jpn. Kokai Tokkyo Koho, 2012126711, 05 Jul 2012
Furumoto, Kentaro et al, Composition containing tetracyclic compound and dissolution aid (4環性化合物を含む組成物), PCT Int. Appl., WO2012023597, 23 Feb 2012, Also published as CA2808210A1, CN103052386A, EP2606886A1, EP2606886A4, US20130143877
Kinoshita, Kazutomo et al,Design and synthesis of a highly selective, orally active and potent anaplastic lymphoma kinase inhibitor (CH5424802), Bioorganic & Medicinal Chemistry, 20(3), 1271-1280; 2012
Kinoshita, Kazutomo et al,9-Substituted 6,6-Dimethyl-11-oxo-6,11-dihydro-5H-benzo[b]carbazoles as Highly Selective and Potent Anaplastic Lymphoma Kinase Inhibitors, Journal of Medicinal Chemistry, 54(18), 6286-6294; 2011
Kinoshita, Kazuhiro et al, Preparation of tetracyclic compounds such as 11-oxo-5,6-dihydrobenzo[b]carbazole-3-carbonitrile derivatives as anaplastic lymphoma kinase (ALK) inhibitors,Jpn. Tokkyo Koho, 4588121, 24 Nov 2010

6 IMATINIB


 Imatinib
Imatinib
CAS No:- [152459-95-5]
IUPAC Name:- 4-[(4-Methyl-1-piperazinyl)methyl]-N-[4-methyl-3-[[4-(3-pyridinyl)-2-pyrimidinyl]amino]phenyl]benzamide
M. P.:- 211-213 °C
MW: 493.604
4-[(4-methylpiperazin-1-yl)methyl]-N-(4-methyl-3-{[4-(pyridin-3-yl)pyrimidin-2-yl]amino}phenyl)benzamide
-[(4-methylpiperazin-1-yl)methyl]-N-(4-methyl-3-{[4-(pyridin-3-yl)pyrimidin-2-yl]amino}phenyl)benzamide
Imatinib (INN), marketed by Novartis as Gleevec (Canada, South Africa and the USA) or Glivec (Australia, Europe and Latin America), and sometimes referred to by its investigational name STI-571, is a tyrosine-kinase inhibitor used in the treatment of multiple cancers, most notably Philadelphia chromosome-positive (Ph+) chronic myelogenous leukemia (CML).[1]
Like all tyrosine-kinase inhibitors, imatinib works by preventing a tyrosine kinase enzyme, in this case BCR-Abl, fromphosphorylating subsequent proteins and initiating the signalling cascade necessary for cancer growth and survival, thus preventing the growth of cancer cells and leading to their death by apoptosis.[2] Because the BCR-Abl tyrosine kinase enzyme exists only in cancer cells and not in healthy cells, imatinib works as a form of targeted therapy—only cancer cells are killed through the drug's action.[3] In this regard, imatinib was one of the first cancer therapies to show the potential for such targeted action, and is often cited as a paradigm for research in cancer therapeutics.[4]
Imatinib has been cited as the first of the exceptionally expensive cancer drugs, costing $92,000 a year. Doctors and patients complain that this is excessive, given that its development costs have been recovered many times over, and that the costs of synthesizing the drug are orders of magnitude lower. In the USA, the patent protecting the active principle will expire on 4 January 2015 while the patent protecting the beta crystal form of the active principal ingredient will expire on 23 May 2019.[5]

bcr-abl kinase (green), which causes CML, inhibited by imatinib (red; small molecule).
Medical uses
Imatinib is used to treat chronic myelogenous leukemia (CML), gastrointestinal stromal tumors (GISTs) and a number of othermalignancies.
Chronic myelogenous leukemia
The U.S. Food and Drug Administration (FDA) has approved imatinib as first-line treatment for Philadelphia chromosome-positive CML, both in adults and children. The drug is approved in multiple Philadelphia chromosome-positive cases of CML, including after stem cell transplant, in blast crisis, and newly diagnosed.[8]
Gastrointestinal stromal tumors
The FDA first granted approval for advanced GIST patients in 2002. On 1 February 2012, imatinib was approved for use after the surgical removal of KIT-positive tumors to help prevent recurrence.[9] The drug is also approved in unresectable KIT-positive GISTs.[8]
Other
The FDA has approved imatinib for use in adult patients with relapsed or refractory Philadelphia chromosome-positive acute lymphoblastic leukemia (Ph+ ALL), myelodysplastic/ myeloproliferative diseases associated with platelet-derived growth factor receptor gene rearrangements, aggressive systemic mastocytosis without or an unknown D816V c-KIT mutation, hypereosinophilic syndrome and/or chronic eosinophilic leukemia who have the FIP1L1-PDGFRα fusion kinase (CHIC2 allele deletion) or FIP1L1-PDGFRα fusion kinase negative or unknown, unresectable, recurrent and/or metastaticdermatofibrosarcoma protuberans.[8] On 25 January 2013, Gleevec was approved for use in children with Ph+ ALL.[10]
For treatment of progressive plexiform neurofibromas associated with neurofibromatosis type I, early research has shown potential for using the c-KIT tyrosine kinase blocking properties of imatinib.[11][12][13][14]
Legal challenge to generics
In 2007, imatinib became a test case through which Novartis challenged India's patent laws. A win for Novartis would make it harder for Indian companies to produce generic versions of drugs still manufactured under patent elsewhere in the world. Doctors Without Borders argues a change in law would make it impossible for Indian companies to produce cheap generic antiretrovirals (anti-HIV medication), thus making it impossible for Third World countries to buy these essential medicines.[43] On 6 August 2007, the Madras High Court dismissed the writ petition filed by Novartis challenging the constitutionality of Section 3(d) of Indian Patent Act, and deferred to the World Trade Organization (WTO) forum to resolve the TRIPS compliance question. As of 2009 India has refused to grant patent exclusivity..
On April 01, 2013 Supreme Court of India dismissed the plea of Novartis for the grant of patent.

in germany
Mechanism of action

| IMATINIB | |
|---|---|
| Drug mechanism | |

Crystallographic structure of tyrosine-protein kinase ABL (rainbow colored, N-terminus = blue, C-terminus = red) complexed with imatinib (spheres, carbon = white, oxygen = red, nitrogen = blue).[31]
| |
| THERAPEUTIC USE | chronic myelogenous leukemia |
| BIOLOGICAL TARGET | ABL, c-kit, PDGF-R |
| MECHANISM OF ACTION | Tyrosine-kinase inhibitor |
| EXTERNAL LINKS | |
| ATC CODE | L01XE01 |
| PDB LIGAND ID | STI: PDBe, RCSB PDB |
| LIGPLOT | 1iep |
Imatinib is a 2-phenyl amino pyrimidine derivative that functions as a specific inhibitor of a number of tyrosine kinase enzymes. It occupies the TK active site, leading to a decrease in activity.
There are a large number of TK enzymes in the body, including the insulin receptor. Imatinib is specific for the TK domain inabl(the Abelson proto-oncogene), c-kit and PDGF-R (platelet-derived growth factorreceptor).
In chronic myelogenous leukemia, the Philadelphia chromosome leads to a fusion protein of abl with bcr(breakpoint cluster region), termed bcr-abl. As this is now aconstitutively active tyrosine kinase, imatinib is used to decrease bcr-abl activity.
The active sites of tyrosine kinases each have a binding site for ATP. The enzymatic activity catalyzed by a tyrosine kinase is the transfer of the terminal phosphate from ATP to tyrosine residues on its substrates, a process known as protein tyrosinephosphorylation. Imatinib works by binding close to the ATP binding site of bcr-abl, locking it in a closed or self-inhibited conformation, and therefore inhibiting the enzyme activity of the protein semi-competitively.[32] This fact explains why many BCR-ABL mutations can cause resistance to imatinib by shifting its equilibrium toward the open or active conformation.[33]
Imatinib is quite selective for bcr-abl – it does also inhibit other targets mentioned above (c-kit and PDGF-R), but no other knowntyrosine kinases. Imatinib also inhibits the abl protein of non-cancer cells but cells normally have additional redundant tyrosine kinases which allow them to continue to function even if abl tyrosine kinase is inhibited. Some tumor cells, however, have a dependence on bcr-abl.[34] Inhibition of the bcr-abl tyrosine kinase also stimulates its entry in to the nucleus, where it is unable to perform any of its normal anti-apoptopic functions.[35]
The Bcr-Abl pathway has many downstream pathways including the Ras/MapK pathway, which leads to increased proliferation due to increased growth factor-independent cell growth. It also affects the Src/Pax/Fak/Rac pathway. This affects the cytoskeleton, which leads to increased cell motility and decreased adhesion. The PI/PI3K/AKT/BCL-2 pathway is also affected. BCL-2 is responsible for keeping the mitochondria stable; this suppresses cell death by apoptosis and increases survival. The last pathway that Bcr-Abl affects is the JAK/STAT pathway, which is responsible for proliferation.[36]
synthesis
..............................
Imatinib is known as an inhibitor of protein-tyrosine kinase and is indicated for the treatment of chronic myeloid leukemia (CML). Imatinib also has potential for the treatment of various other cancers that express these kinase including acute lymphocyte leukemia and certain solid tumors. It can also be used for the treatment of atherosclerosis, thrombosis, restenosis, or fibrosis. Thus, imatinib can also be used for the treatment of non-malignant diseases. Imatinib is usually administered orally in the form of a suitable salt, e.g., in the form of imatinib mesylate.
The chemical name of Imatinib is 4-(4-methyl piperazine -1- methyl) -N-4-methyl-3-[4- (3- pyridyl) pyrimidine-2-amino] - benzamide and is represented by the following structural formula:

(Imatinib)
Imatinib Mesylate is an inhibitor of signal transduction (STI571) invented by Novartis AG after 7 years of hard work; it is the first inhibitor of cancer signal transduction ratified in the whole world. It is sold by Novartis as Gleevec capsules containing imatinib mesylate in amounts equivalent to 100 mg or 400 mg of imatinib free base.
Imatinib Mesylate is the rare drug in America, European Union and Japan. In May 10, 2001, it was ratified by American Food and Drug Administration (FDA) to treat the chronic myelogenous leukemia patients. EP0564409 (US5521 184) describes the process for the preparation of imatinib and the use thereof, especially as an anti tumour agent.
There are generally two synthetic routes for synthesis of Imatinib, suitable for the industrial production. One synthetic process as described in scheme-I comprises using 2-methyl-5-nitroaniline as the raw material which is reacted with cyanamide to obtain guanidine; cyclization reaction with 3-dimethylamino-l-(3-pyridyl)-2-propylene-l- ketone; reduction step of nitro to amine and condensation reaction with 4- (Chloromethyl)benzoyl chloride and N-methylpiperazidine to obtain Imatinib (WO 2004/108669). -I

Scheme-2 describes the successful process for the synthesis of Imatinib using 4-methyl-3- nitroanilines as the raw material, comprising reacting 4-methyl-3-nitroanilines with 4- (Chloromethyl)benzoyl chloride and N-methyl piperazidine in turns; followed by reduction of nitro group to amino group; then reaction with cyanamide to obtain guanidine; finally cyclization reaction with 3- dimethyl amino- 1 -(3- pyridyl)-2- propylene-1 -ketone to obtain Imatinib (WO 03/066613). The said PCT application discloses the use of 4-4-(methyl piperazin-l-ylmethyl)-benzoic acid methyl ester as one of the raw material but rest of the reactants are different from that of N-(5-amino -2- methylphenyl)-4-(3-pyridyl)-2-pyrimidine amine in presence of trimethyl aluminium.
Scheme-2

Common feature of the processes for preparing imatinib according to (WO 2004/108669) and (WO03/066613) lies in use of cyanamide as a reagent. The main difference between the two routes is that the reaction sequence of cyclization of pyrimidine chain is different. Example 10 of PCT International Publication no. WO 2003/066613 is less applicable to industrial purposes. These include the reaction between N-(3-bromo-4-methyl-phenyl)-4- (4-methyl-piperazin-l -ylmethyl)-benzamide and 4-(3-pyridyl)-2-pyrimidineamine which uses a mixture of rac-BINAP (a phosphine oxide catalyst) and Pd2 (dba)3*CHCl3. These catalysts are very expensive, therefore, their use is unfit for commercial production.
CN1630648A describes a process comprising reaction of 3- bromine-4- methyl aniline with 4-(4-methyl-piperazin- methyl) methyl benzoate in presence of trimethyl-Aluminum to obtain N-(4-methyl-3-bromobenzene)-4-(4-methyl-piperazin- 1 -methyl)-benzamide, which further reacts with 2-amino-4-(3-pyridyl)- pyrimidine in presence of palladium as catalyst to obtain Imatinib.


The drawback of the above process is the use of trimethyl-Aluminum, which is flammable and reacts severely when comes in contact with water.
CN101016293A describes another process using N-(4-methyl-3-3- aminophenyl)-4-(4- methyl-piperazin-1 -methyl)- benzamide as the raw material. The said raw material is reacted with 2-halogen-4-(3-pyridyl)- pyrimidine to obtain Imatinib.

The process disclosed in CN 101016293 A comprises use of halogenated agent, such as phosphorus oxychloride, which is used to synthesize 2-halogeno-4- methyl- (3-pyridyl) - pyridine is lachrymator and corrosive and has great influence to the surroundings. EP0564409 describes a coupling reaction between N-(5-amino -2-methylphenyl)-4-(3- pyridyl)-2-pyrimidine amine and 4-(4-methyl piperazin-l-ylmethyl)-benzoyl chloride in the presence of high quantity of pyridine to starting reactant amine N-(5-amino -2- methylphenyl)-4-(3-pyridyl)-2-pyrimidine amine. The ratio of the pyridine to the said reactant is 138 which is equivalent to about 40 parts v/w. Use of such a large quantity of pyridine is unsafe as it is a toxic solvent according to ICH guidelines. The workup of the reaction comprises evaporation of the remaining pyridine and further processing, which finally involves chromatography for purification, which is highly undesirable on industrial scale because it is expensive and time consuming.

US2006/0149061 and US20060223817 also discloses a similar synthetic approach comprising the use of similar pyridine /starting amine ratio (140 equivalents which is equals about 41 parts v/w). The product obtained is purified by slurring in ethyl acetate.
WO2004/074502 describes a coupling reaction between N-(5-amino -2-methylphenyl)-4- (3-pyridyl)-2-pyrimidine amine and 4-(4-methyl piperazin-l-ylmethyl)-benzoyl chloride wherein solvent like dimethyl pharmamide , dimethyl acetamide, N-methyl pyrilidinone are used as solvents instead of pyridine. However the method described in this patent application lacks an advantage in that the coupling reaction produces the hydrohalide salt of imatinib, e.g. imatinib trihydrochloride monohydrate, which has to be treated with a base in order to afford the imatinib base, thus an extra step is required. Further, conventional methods for coupling N-(5-amino -2-methylphenyl)-4-(3-pyridyl)-2- pyrimidine amine require reaction with an acid halide, e.g. 4-(4-methyl piperazin-1- ylmethyl)-benzoyl chloride, which requires an additional production step that can involve harsh and/or toxic chlorinating agent.

WO2008/1 17298 describes a coupling reaction between N-(5-amino -2-methylphenyl)-4- (3-pyridyl)-2-pyrimidine amine and 4-(4-methyl piperazin-l-ylmethyl)-benzoyl chloride in presence of a base selected from potassium carbonate, sodium carbonate, potassium or sodium hydroxide. Use of potassium carbonate as base results into the formation of Imatinib dihydrochloride which ultimately requires an additional operation of neutralization by using excessive base to get imatinib.

WO2008/136010 describes a coupling reaction between N-(5-amino -2-methylphenyl)-4- (3-pyridyl)-2-pyrimidine amine and 4-(4-methyl piperazin-l-ylmethyl)-benzoyl chloride in presence of base potassium hydroxide resulting into 78.6% yield of crude imatinib base. Preparation of crude requires imatinib hydrochloride preparation during the workup which is then basified to get base in crude form. This also describes maleate salt preparation as mode of purification which is again basified to give pure Imatinib base.

US patent application 2004/0248918 discloses a different approach using N-(5-amino -2- methylphenyl)-4-(3-pyridyl)-2-pyrimidine amine and 4-(2-chloromethyl)-benzoyl chloride as raw material. The reaction for the preparation of Imatinib is carried out in tetrahydrofuran as a reaction solvent and in the presence of pyridine as a base. However the method described in this patent application lacks an advantage as purification of the product requires column chromatography using chloroform: methanol (3: 1 v/v), which is not suitable purification method when performing the reaction on large scale, followed by crystallizati

US patent application 2008/0103305 discloses a process comprising reacting N-(5-amino -2-methylphenyl)-4-(3-pyridyl)-2-pyrimidine amine or its alkyl derivative and an acid salt of 4-[(4-methyl-l-piperazinyl)-methyl] benzoyl derivative as given below in the scheme-3 using pyridine in an amount of about 2 to 10 volumes per gram of the said amine. However the drawback associated with this process is use of pyridine especially when reaction is performed on large scale. -3

...............................
SYNTHESIS
")
Inverse synthetic analysis will be divided into four imatinib into fragment A has 1,3 - parents electrical, fragment B are 1,3 - parent nuclear, fragments A and B constitute a pyrimidine ring.
")
Compound 4 can be obtained in two ways, benzyl bromide 1 and secondary amines 2 by SN2 reaction, or the aldehyde 3 with a secondary amine 2 by reductive amination. Sodium cyanoborohydride electron withdrawing effect of the cyano group, thereby reducing the activity of the negative hydrogen, it may be present in acidic solution. Also in the acidic conditions of aldehydes and secondary amines imine positive ions, which is higher than the activity of aldehyde reduction.This is why the reductive amination reagent with inert negative and hydrogen under acidic conditions. 4 hydrolyzed ester with thionyl chloride into the acid chloride 5 . The reaction of aniline and cyanamide dinucleophile guanidine 7 . Compound 8 and DMF-DMA reaction electrophilic reagent parents 9 , 7 , and 9 ring closure under alkaline conditions to generate 10 . Finally, reduction, amidation, and a salt of imatinib mesylate generated.
......................................
Org. Process Res. Dev., 2012, 16 (11), pp 1794–1804
DOI: 10.1021/op300212u

An efficient, economic process has been developed for the production of imatinib with 99.99% purity and 50% overall yield from four steps. Formation and control of all possible impurities is described. The synthesis comprises the condensation of N-(5-amino-2-methylphenyl)-4-(3-pyridinyl)-2-pyrimidineamine with 4-(4-methylpiperazinomethyl)benzoyl chloride in isopropyl alcohol solvent in the presence of potassium carbonate to yield imatinib base.
..............................
Org. Biomol. Chem., 2013,11, 1766-1800
DOI: 10.1039/C2OB27003J


Imatinib (1), nilotinib (2) and dasatinib (3) are Bcr-Abl tyrosine kinase inhibitors approved for the treatment of chronic myelogenous leukemia (CML). This review collates information from the journal and patent literature to provide a comprehensive reference source of the different synthetic methods used to prepare the aforementioned active pharmaceutical ingredients (API's).

..........................
|









![[1860-5397-9-265-i37]](http://www.beilstein-journals.org/bjoc/content/inline/1860-5397-9-265-i37.png?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-9-265-i38]](http://www.beilstein-journals.org/bjoc/content/inline/1860-5397-9-265-i38.png?scale=2.0&max-width=1024&background=FFFFFF)



Medicine for Blood Cancer
‘Imitinef Mercilet’ is a medicine which cures blood cancer.
Its available free of cost at “Adyar Cancer Institute in Chennai”.
Create Awareness. It might help someone.Cancer Institute in Adyar, Chennai
Its available free of cost at “Adyar Cancer Institute in Chennai”.
Create Awareness. It might help someone.Cancer Institute in Adyar, Chennai
‘Imitinef Mercilet’ is apparently an alternative spelling of the drug Imatinib mesylate which is used in the treatment of some forms of leukemia along with other types of cancer. Imatinib, often referred to a “Gleevec”, has proved to be an effective treatment for some forms of cancers. However, “blood cancer” is a generalized term for cancers that affect the blood, lymphatic system or bone marrow. The three types of blood cancer are listed as leukemia, lymphoma, and multiple myeloma. These three malignancies require quite different kinds of treatments. While drugs (including Imatinib), along with other treatments such as radiation can help to slow or even stop the progress of these cancers, there is currently no single drug treatment that can be said to actually cure all such cancers.
Category: Cancer
Address: East Canal Bank Road , Gandhi Nagar
Adyar, Chennai -600020
Landmark: Near Michael School
Phone: 044-24910754 044-24910754 ,
044-24911526 044-24911526 , 044-22350241
Address: East Canal Bank Road , Gandhi Nagar
Adyar, Chennai -600020
Landmark: Near Michael School
Phone: 044-24910754 044-24910754 ,
044-24911526 044-24911526 , 044-22350241
Imatinib is a small molecule selectively inhibiting specific tyrosine kinases that has emerged recently as a valuable treatment for patients with advanced GIST. The use of imatinib as monotherapy for the treatment of GIST has been described in PCT publication WO 02/34727, which is here incorporated by reference. However, it has been reported that primary resistance to imatinib is present in a population of patients, for example 13.7% of patients in one study. In addition, a number of patients acquire resistance to treatment with imatinib. More generally this resistance is partial with progression in some lesions, but continuing disease control in other lesions. Hence, these patients remain on imatinib treatment but with a clear need for additional or alternative therapy.
Imatinib is 4-(4-methylpiperazin-1-ylmethyl)-N-[4-methyl-3-(4-pyridin-3-yl)pyrimidin-2-ylamino)phenyl]-benzamide having the formula I

The preparation of imatinib and the use thereof, especially as an anti-tumour agent, are described in Example 21 of European patent application EP-A-0 564 409, which was published on 6 Oct. 1993, and in equivalent applications and patents in numerous other countries, e.g. in U.S. Pat. No. 5,521,184 and in Japanese patent 2706682

The flow-based route required minimal manual intervention and was achieved despite poor solubility of many reaction components
21 January 2013Michael Parkin
UK chemists have used a combination of flow chemistry methods with solid-supported scavengers and reagents to synthesise the active pharmaceutical ingredient, imatinib, of the anticancer drug Gleevec. The method avoids the need for any manual handling of intermediates and allows the drug to be synthesised in high purity in less than a day.
Gleevec, developed by Novartis, is a tyrosine kinase inhibitor used for the treatment of chronic myeloid leukaemia and gastrointestinal stromal tumours.

READ ALL AT

 | |
|---|---|
 | |
| IMATINIB |

CREDIT

'Wrapping' Gleevec Fights Drug-Resistant Cancer, Study Shows
The anti-cancer drug Gleevec® is far more effective against a drug-resistant strain of cancer when the drug wraps the target with a molecular bandage that seals out water from a critical area. This image shows the bandage (black box) on the modified version of the drug, WBZ-7. (Credit: Image courtesy of Rice University)
A new study in Cancer Research finds that the anti-cancer drug Gleevec® is far more effective against a drug-resistant strain of cancer when the drug wraps the target with a molecular bandage that seals out water from a critical area.


FIG 23.8 Optimization of imatinib as a chemotherapeutic agent. The discovery that 2-phenylaminopyrimidine inhibitors of PKC also inhibit the unrelated v-Abl oncogene turned attention to its potential use in the treatment of chronic myelogenous leukaemia. Starting with the 2-phenylaminopyrimidine backbone, addition of the benzamidine group increased activity against tyrosine kinases, the methyl group reduced its activity against PKC (so-called ‘ target hopping ’ ). Addition of a 3’-pyridyl group improved the activity in cellular assays. Subsequent addition of N -methylpiperazine increased water solubility and oral bioavailability, enabling the drug to survive the stomach and to enter the bloodstream.
..........................
An automated flow-based synthesis of imatinib: the API of gleevec M.D. Hopkin, I.R. Baxendale, S.V. Ley, J.C.S. Chem. Commun.2010, 46, 2450-2452.

References
- Jump up^ Novartis Pharma AG. Gleevec® (imatinib mesylate) tablets prescribing information. East Hanover, NJ; 2006 Sep. Anon. Drugs of choice for cancer. Treat Guidel Med Lett. 2003; 1:41–52
- Jump up^ Goldman JM, Melo JV (October 2003). "Chronic myeloid leukemia--advances in biology and new approaches to treatment". N. Engl. J. Med. 349 (15): 1451–64.doi:10.1056/NEJMra020777. PMID 14534339.
- Jump up^ Fausel, C. Targeted chronic myeloid leukemia therapy: Seeking a cure. Am J Health Syst Pharm 64, S9-15 (2007)
- Jump up^ Stegmeier F, Warmuth M, Sellers WR, Dorsch M (May 2010). "Targeted cancer therapies in the twenty-first century: lessons from imatinib". Clin. Pharmacol. Ther. 87(5): 543–52. doi:10.1038/clpt.2009.297. PMID 20237469.
- Jump up^ "Novartis fails to patent Glivec (Gleevec) in India".
- Jump up^ Rowley to receive Japan Prize for her role in the development of targeted cancer therapy Eurekalert, Press release, 24 January 2012
- Jump up^ Leukemia Drug and Magnet Material Net Japan Prizes by Dennis Normile, Science Insider, 25 January 2012
- ^ Jump up to:a b c "FDA Highlights and Prescribing Information for Gleevec(imatinib mesylate)".
- Jump up^ "Prolonged Use of Imatinib in GIST Patients Leads to New FDA Approval".
- Jump up^ "FDA approves Gleevec for children with acute lymphoblastic leukemia". FDA News Release. US Food and Drug Administration. 25 January 2013. Retrieved 3 April 2013.
- Jump up^ Yang FC, Ingram DA, Chen S, Zhu Y, Yuan J, Li X, Yang X, Knowles S, Horn W, Li Y, Zhang S, Yang Y, Vakili ST, Yu M, Burns D, Robertson K, Hutchins G, Parada LF, Clapp DW (October 2008). "Nf1-dependent tumors require a microenvironment containing Nf1+/--and c-kit-dependent bone marrow". Cell 135 (3): 437–48.doi:10.1016/j.cell.2008.08.041. PMC 2788814. PMID 18984156. Lay summary –Science Daily.
- Jump up^ "Gleevec NF1 Trial". Nfcure.org. Retrieved 2013-04-03.
- Jump up^ "GIST in Neurofibromatosis 1". Gistsupport.org. 2010-05-14. Retrieved 2013-04-03.
- Jump up^ ""Pilot Study of Gleevec/Imatinib Mesylate (STI-571, NSC 716051) in Neurofibromatosis (NF1) Patient With Plexiform Neurofibromas (0908-09)" (Suspended)". Clinicaltrials.gov. Retrieved 2013-04-03.
- Jump up^ Droogendijk HJ, Kluin-Nelemans HJ, van Doormaal JJ, Oranje AP, van de Loosdrecht AA, van Daele PL (July 2006). "Imatinib mesylate in the treatment of systemic mastocytosis: a phase II trial". Cancer 107 (2): 345–51. doi:10.1002/cncr.21996.PMID 16779792.
- Jump up^ Tapper EB, Knowles D, Heffron T, Lawrence EC, Csete M (June 2009). "Portopulmonary hypertension: imatinib as a novel treatment and the Emory experience with this condition". Transplant. Proc. 41 (5): 1969–71.doi:10.1016/j.transproceed.2009.02.100. PMID 19545770.
- Jump up^ Boucher P, Gotthardt M, Li WP, Anderson RG, Herz J (April 2003). "LRP: role in vascular wall integrity and protection from atherosclerosis". Science 300 (5617): 329–32.doi:10.1126/science.1082095. PMID 12690199.
- Jump up^ Lassila M, Allen TJ, Cao Z, Thallas V, Jandeleit-Dahm KA, Candido R, Cooper ME (May 2004). "Imatinib attenuates diabetes-associated atherosclerosis". Arterioscler. Thromb. Vasc. Biol. 24 (5): 935–42. doi:10.1161/01.ATV.0000124105.39900.db.PMID 14988091.
- Jump up^ Reeves PM, Bommarius B, Lebeis S, McNulty S, Christensen J, Swimm A, Chahroudi A, Chavan R, Feinberg MB, Veach D, Bornmann W, Sherman M, Kalman D (July 2005). "Disabling poxvirus pathogenesis by inhibition of Abl-family tyrosine kinases". Nat. Med.11 (7): 731–9. doi:10.1038/nm1265. PMID 15980865.
- Jump up^ He G, Luo W, Li P, Remmers C, Netzer WJ, Hendrick J, Bettayeb K, Flajolet M, Gorelick F, Wennogle LP, Greengard P (September 2010). "Gamma-secretase activating protein is a therapeutic target for Alzheimer's disease". Nature 467 (7311): 95–8.doi:10.1038/nature09325. PMC 2936959. PMID 20811458.
- Jump up^ "Alzheimer's may start in liver – Health – Alzheimer's Disease | NBC News". MSNBC. Retrieved 2013-01-06.
- Jump up^ Holmes C, Boche D, Wilkinson D, Yadegarfar G, Hopkins V, Bayer A, Jones RW, Bullock R, Love S, Neal JW, Zotova E, Nicoll JA (July 2008). "Long-term effects of Abeta42 immunisation in Alzheimer's disease: follow-up of a randomised, placebo-controlled phase I trial". Lancet 372 (9634): 216–23. doi:10.1016/S0140-6736(08)61075-2. PMID 18640458.
- Jump up^ Eliminating Morphine Tolerance – Reformulated Imatinib 23 Feb 2012, 5:00 PST
- Jump up^ "GLIVEC Tablets - Summary of Product Characteristics (SPC)". electronic Medicines Compendium. Novartis Pharmaceuticals UK Ltd.
- ^ Jump up to:a b c "Gleevec (imatinib) dosing, indications, interactions, adverse effects, and more".Medscape Reference. WebMD. Retrieved 24 January 2014.
- Jump up^ "Imatinib". Macmillan Cancer Support. Retrieved 26 December 2012.
- ^ Jump up to:a b Haberfeld, H, ed. (2009). Austria-Codex (in German) (2009/2010 ed.). Vienna: Österreichischer Apothekerverlag. ISBN 3-85200-196-X.
- Jump up^ Kerkelä R, Grazette L, Yacobi R, Iliescu C, Patten R, Beahm C, Walters B, Shevtsov S, Pesant S, Clubb FJ, Rosenzweig A, Salomon RN, Van Etten RA, Alroy J, Durand JB, Force T (August 2006). "Cardiotoxicity of the cancer therapeutic agent imatinib mesylate". Nat. Med. 12 (8): 908–16. doi:10.1038/nm1446. PMID 16862153.
- Jump up^ Shima H, Tokuyama M, Tanizawa A, Tono C, Hamamoto K, Muramatsu H, Watanabe A, Hotta N, Ito M, Kurosawa H, Kato K, Tsurusawa M, Horibe K, Shimada H (October 2011). "Distinct impact of imatinib on growth at prepubertal and pubertal ages of children with chronic myeloid leukemia". J. Pediatr. 159 (4): 676–81.doi:10.1016/j.jpeds.2011.03.046. PMID 21592517.
- ^ Jump up to:a b c d "GLIVEC (imatinib)" (PDF). TGA eBusiness Services. Novartis Pharmaceuticals Australia Pty Ltd. 21 August 2013. Retrieved 24 January 2014.
- Jump up^ PDB 1IEP; Nagar B, Bornmann WG, Pellicena P, Schindler T, Veach DR, Miller WT, Clarkson B, Kuriyan J (August 2002). "Crystal structures of the kinase domain of c-Abl in complex with the small molecule inhibitors PD173955 and imatinib (STI-571)". Cancer Res. 62 (15): 4236–43. PMID 12154025.
- Jump up^ Takimoto CH, Calvo E. "Principles of Oncologic Pharmacotherapy" in Pazdur R, Wagman LD, Camphausen KA, Hoskins WJ (Eds)Cancer Management: A Multidisciplinary Approach. 11 ed. 2008.
- Jump up^ Gambacorti-Passerini CB, Gunby RH, Piazza R, Galietta A, Rostagno R, Scapozza L (February 2003). "Molecular mechanisms of resistance to imatinib in Philadelphia-chromosome-positive leukaemias". Lancet Oncol. 4 (2): 75–85. doi:10.1016/S1470-2045(03)00979-3. PMID 12573349.
- Jump up^ Deininger MW, Druker BJ (September 2003). "Specific targeted therapy of chronic myelogenous leukemia with imatinib". Pharmacol. Rev. 55 (3): 401–23.doi:10.1124/pr.55.3.4. PMID 12869662.
- Jump up^ Vigneri P, Wang JY (February 2001). "Induction of apoptosis in chronic myelogenous leukemia cells through nuclear entrapment of BCR-ABL tyrosine kinase". Nat. Med. 7 (2): 228–34. doi:10.1038/84683. PMID 11175855.
- Jump up^ Weisberg E, Manley PW, Cowan-Jacob SW, Hochhaus A, Griffin JD (May 2007). "Second generation inhibitors of BCR-ABL for the treatment of imatinib-resistant chronic myeloid leukaemia". Nature Reviews Cancer 7 (5): 345–56. doi:10.1038/nrc2126.PMID 17457302.
- Jump up^ Scheinfeld N, Schienfeld N (February 2006). "A comprehensive review of imatinib mesylate (Gleevec) for dermatological diseases". J Drugs Dermatol 5 (2): 117–22.PMID 16485879.
- Jump up^ Klopp, T, ed. (2010). Arzneimittel-Interaktionen (in German) (2010/2011 ed.). Arbeitsgemeinschaft für Pharmazeutische Information. ISBN 978-3-85200-207-1.
- ^ Jump up to:a b Staff, Innovation.org (a project of the Pharmaceutical Research and Manufacturers of America)The Story of Gleevec
- Jump up^ Claudia Dreifus for the New York Times. November 2, 2009 Researcher Behind the Drug Gleevec
- ^ Jump up to:a b A Conversation With Brian J. Druker, M.D., Researcher Behind the Drug Gleevecby Claudia Dreifus, The New York Times, 2 November 2009
- Jump up^ Gambacorti-Passerini C (2008). "Part I: Milestones in personalised medicine—imatinib". Lancet Oncology 9 (600): 600. doi:10.1016/S1470-2045(08)70152-9.PMID 18510992.
- Jump up^ Druker BJ, Lydon NB (January 2000). "Lessons learned from the development of an abl tyrosine kinase inhibitor for chronic myelogenous leukemia". J. Clin. Invest. 105 (1): 3–7. doi:10.1172/JCI9083. PMC 382593. PMID 10619854.
- ^ Jump up to:a b c U.S. Patent 5,521,184
- Jump up^ "Imatinib Patent Family". Espacenet. 1996. Retrieved 2014-07-23.
- ^ Jump up to:a b EP 0564409
- Jump up^ Staff, European Medicines Agency, 2004.EMEA Scientific Discussion of Glivec
- Jump up^ Note: The Indian patent application, which became the subject of litigation in India that gathered a lot of press, does not appear to be publicly available. However according todocuments produced in the course of that litigation (page 27), "The Appellant’s application under the PCT was substantially on the same invention as had been made in India."
- ^ Jump up to:a b WO 9903854
- Jump up^ U.S. Patent 6,894,051
- Jump up^ FDA Orange Book; Patent and Exclusivity Search Results from query on Appl No 021588 Product 001 in the OB_Rx list.
- Jump up^ Novartis press release, May 10, 2001. [http://www.evaluategroup.com/Universal/View.aspx?type=Story&id=5838 FDA approves Novartis' unique cancer medication Glivec®
- Jump up^ Cohen MH et al. Approval Summary for Imatinib Mesylate Capsules in the Treatment of Chronic Myelogenous Leukemia Clin Cancer Res May 2002 8; 935
- Jump up^ Margot J. Fromer for Oncology Times. December 2002. What’s in a Name? Quite a Lot When It Comes to Marketing & Selling New Cancer Drugs
- Jump up^ Novartis Press Release. April 30 2001Novartis Oncology Changes Trade Name of Investigational Agent Glivec(TM) to Gleevec(TM) in the United States
- Jump up^ Experts in Chronic Myeloid Leukemia. The price of drugs for chronic myeloid leukemia (CML) is a reflection of the unsustainable prices of cancer drugs: from the perspective of a large group of CML experts Blood. 2013 May 30;121(22):4439-42. PMID 23620577
- Jump up^ Andrew Pollack for the New York Times, April 25, 2013 Doctors Denounce Cancer Drug Prices of $100,000 a Year
- Jump up^ Schiffer CA (July 2007). "BCR-ABL tyrosine kinase inhibitors for chronic myelogenous leukemia". N. Engl. J. Med. 357 (3): 258–65. doi:10.1056/NEJMct071828.PMID 17634461.
- Jump up^ As Pills Treat Cancer, Insurance Lags Behind, By ANDREW POLLACK, New York Times, 14 April 2009
- Jump up^ Living With a Formerly fatal Blood Cancer, By JANE E. BRODY, New York Times, 18 January 2010
- Jump up^ Patented Medicine Review Board (Canada). Report on New Patented Drugs – Gleevec.
- Jump up^ "pharmacychecker.com". pharmacychecker.com. Retrieved 2013-04-03.
- Jump up^ Gardiner Harris and Katie Thomas for the New York Times. April 1 2013 Top court in India rejects Novartis drug patent
- Jump up^ Note: The Indian patent application No.1602/MAS/1998 does not appear to be publicly available. However according to the decision of the IPAB on 26 June 2009 (page 27) discussed below, "The Appellant’s application under the PCT was substantially on the same invention as had been made in India."
- Jump up^ Staff, European Medicines Agency, 2004. EMEA Scientific Discussion of Glivec
- Jump up^ Indian Supreme Court Decision paragraphs 5-6
- Jump up^ Novartis v UoI, para 8-9
- ^ Jump up to:a b Shamnad Basheer for Spicy IP March 11, 2006First Mailbox Opposition (Gleevec) Decided in India
- Jump up^ Staff, LawyersCollective. September 6, 2011[http://www.lawyerscollective.org/news/archived-news-a-articles/126-novartis-case-background-and-update-supreme-court-of-india-to-recommence-hearing.html Novartis case: background and update – Supreme Court of India to recommence hearing
- Jump up^ R. Jai Krishna and Jeanne Whalen for the Wall Street Journal. April 1, 2013Novartis Loses Glivec Patent Battle in India
- Jump up^ Intellectual Property Appellate Board decision dated 26 June 2009, p 149
- Jump up^ W.P. No.24759 of 2006
- Jump up^ "Supreme Court rejects bid by Novartis to patent Glivec".
- Jump up^ Novartis v UoI, Para 191
- Jump up^ Novartis v UoI, Para 24-25
- Jump up^ "How the Indian judgment will reverberate across the world".
- Jump up^ "Patented drugs must be priced smartly".
- Jump up^ Patent with a purpose, Prof. Shamnad Basheer, Indian Express, 3 April 2013
- Kevin Grogan for PharmaTimes. February 27, 2012 Novartis explains stance over India patent law challenge
- Berne Declaration. May 8, 2007 Short questions and answers about the court case initiated by Novartis in India
External links

Title: Imatinib
CAS Registry Number: 152459-95-5
CAS Name: 4-[(4-Methyl-1-piperazinyl)methyl]-N-[4-methyl-3-[[4-(3-pyridinyl)-2-pyrimidinyl]amino]phenyl]benzamide
Additional Names: N-[5-[4-(4-methylpiperazinomethyl)benzoylamido]-2-methylphenyl]-4-(3-pyridyl)-2-pyrimidineamine
Molecular Formula: C29H31N7O
Molecular Weight: 493.60
Percent Composition: C 70.57%, H 6.33%, N 19.86%, O 3.24%
Literature References: Tyrosine kinase inhibitor; highly specific for BCR-ABL, the enzyme associated with chronic myelogenous leukemia (CML) and certain forms of acute lymphoblastic leukemia (ALL). Also shown to inhibit the transmembrane receptor KIT and platelet-derived growth factor (PDGF) receptors. Prepn: J. Zimmermann, EP 564409; idem, US 5521184 (1993, 1996 both to Ciba-Geigy); idem et al., Bioorg. Med. Chem. Lett. 7, 187 (1997). Structural mechanism of ABL specificity: T. Schindler et al., Science 289, 1938 (2000). Activity vs KIT and PDGF receptor kinases: E. Buchdunger et al., J. Pharmacol. Exp. Ther. 295, 139 (2000). Clinical trial in CML: H. Kantarjian et al., N. Engl. J. Med. 346, 645 (2002); in gastrointestinal stromal tumors related to KIT: G. D. Demetri et al., ibid. 347, 472 (2002). Review of clinical experience: D. G. Savage, K. H. Antman, ibid. 346, 683-693 (2002); and pharmacology: V. K. Pindolia et al., Pharmacotherapy 22, 1249-1265 (2002); and development of therapeutic target: B. J. Druker, Adv. Cancer Res. 91, 1-30 (2004).
Properties: mp 211-213°. pKa1 8.07; pKa2 3.73; pKa3 2.56; pKa4 1.52.
Melting point: mp 211-213°
pKa: pKa1 8.07; pKa2 3.73; pKa3 2.56; pKa4 1.52

Derivative Type: Methanesulfonate
CAS Registry Number: 220127-57-1
Manufacturers' Codes: STI-571; CGP-57148B
Trademarks: Gleevec (Novartis); Glivec (Novartis)
Molecular Formula: C29H31N7O.CH3SO3H
Molecular Weight: 589.71
Percent Composition: C 61.10%, H 5.98%, N 16.63%, O 10.85%, S 5.44%
Literature References: Prepn of crystalline form: J. Zimmermann et al., WO 9903854 (1999 to Novartis).
Properties: Occurs in 2 crystalline modifications. a-form, begins to melt at 226°; b-form, mp 217°. Lipophilic at pH 7.4. Soly in water: >100 g/l (pH 4.2); 49 mg/l (pH 7.4).
Melting point: mp 217°
Therap-Cat: Antineoplastic.
Keywords: Antineoplastic, Tyrosine Kinase Inhibitors, imatinib mesylate, GGP-57148B, STI-571, CGP-57148 (free base), Gleevec, Glivec, imatinib
7
NERATINIB






















































NERATINIB
NERATINIB
(2E)-N-[4-[[3-chloro-4-[(pyridin-2-yl)methoxy]phenyl]amino]-3-cyano-7-ethoxyquinolin-6-yl]-4-(dimethylamino)but-2-enamide
[(2E)-N-[4-[[3-chloro-4- [(pyridin-2-yl)methoxy]phenyl]amino]-3-cyano-7-ethoxyquinolin-6-yl]-4- (dimethylamino)but-2-enamide].
(E)-N- {4-[3-chloro-4-(2-pyridinylmethoxy)anilino]-3-cyano-7-ethoxy-6- quinolinyl} -4-(dimethylamino)-2-butenamide
FOR METASTATIC BREAST CANCER.PHASE 3
CAS 698387-09-6,
PFIZER …….INNOVATOR
HKI-272, HKI 272, Neratinib(HKI-272), Neratinib, HKI-272, 698387-09-6, HKI272, HKI 272, HKI-272,
HKI-272
PB-272
PF-0528767
WAY-179272
WAY-179272-B (maleate)
PB-272
PF-0528767
WAY-179272
WAY-179272-B (maleate)
Molecular Formula: C30H29ClN6O3
Molecular Weight: 557.04266
Puma Biotechnology, a development stage biopharmaceutical company, announced the presentation of positive results from the phase II clinical trial of Puma’s investigational drug PB272 (neratinib) for the neoadjuvant treatment of breast cancer(I-SPY 2 TRIAL) in an oral presentation at the American Association for Cancer Research (AACR) Annual Meeting 2014 in San Diego, California.
READ AT
http://www.pharmabiz.com/NewsDetails.aspx?aid=81352&sid=2 …….COPY PASTE LINK
| Neratinib – малая молекула класса 6,7-дизамещенных-4-anilinoquinoline-3-карбонитрила - ингибитор тирозинкиназы HER-2 с потенциальной противоопухолевой активностью. Neratinib связывается с рецептором HER-2 необратимо, снижая аутофосфорилирование в клетках, и направляя остаток цистеина в АТФ-связывающего кармана рецептора. Обработка раковых клеток с этим агентом приводит к торможению передачи сигнала клеточного цикла и в конечном счете уменьшает клеточную пролиферацию. Neratinib ингибирует рецептор EGFR киназы и распространение EGFR-зависимых клеток. | |
| Neratinib – small molecule 6,7-disubstituted class of 4-anilinoquinoline-3-carbonitrile - inhibitor of the HER-2 tyrosine kinase with potential antitumor activity. Neratinib binds to the receptor HER-2 irreversible, reducing autophosphorylation in cells and directing the cysteine residue in the ATP-binding pocket of the receptor. Treatment of cancer cells with this agent leads to inhibition of signal transduction and cell cycle ultimately reducescell proliferation. Neratinib inhibit EGFR kinase receptor and distribution of EGFR-dependent cells. |
Neratinib (HKI-272) is a tyrosine kinase inhibitor[1][2] under investigation for the treatment breast cancer[3] and other solid tumours.
Like lapatinib and afatinib, it is a dual inhibitor of the human epidermal growth factor receptor 2 (Her2) and epidermal growth factor receptor (EGFR) kinases.[5]
Neratinib is a signal transduction pathway inhibitor and an irreversible inhibitor of HER-2 in early clinical trials for the treatment of advanced solid tumors in combination with paclitaxel. The company had also been developing the drug candidate for the treatment of non-small cell lung cancer (NSCLC); however, no recent development has been reported for the indication. In 2011, Pfizer discontinued development of the compound as monotherapy for the treatment of ErbB-2-positive breast cancer. A phase III clinical trial had been under way. Dana-Farber Cancer Institute is studying the compound for the treatment of patients with human epidermal growth factor receptor 2 (HER2)-positive breast cancer and brain metastases. Puma Biotechnology is conducting phase III trials for use as third-line treatment of HER2-positive metastatic breast cancer and phase II trials for the treatment of patients with HER2 activating mutations in Non-Small Cell Lung Cancer (as monotherapy or in combination with temsirolimus) as well as other solid tumors.
The drug candidate is a synthetic compound developed based on the chemical structure of EKB-569, an inhibitor of the epidermal growth factor receptor (EGFR) currently under clinical evaluation for the treatment of EGFR-positive tumors. In previous trials, neratinib inhibited kinase activity of HER-2 and EGFR by 50% while showing no effects on several serine-threonine kinases, and also inhibited the proliferation of two HER-2-positive breast cancer cell lines and a mouse fibroblast cell line transfected with the HER-2 oncogene.
In 2011, the compound was licensed to Puma by Pfizer for global development and commercialization.
The drug candidate is a synthetic compound developed based on the chemical structure of EKB-569, an inhibitor of the epidermal growth factor receptor (EGFR) currently under clinical evaluation for the treatment of EGFR-positive tumors. In previous trials, neratinib inhibited kinase activity of HER-2 and EGFR by 50% while showing no effects on several serine-threonine kinases, and also inhibited the proliferation of two HER-2-positive breast cancer cell lines and a mouse fibroblast cell line transfected with the HER-2 oncogene.
In 2011, the compound was licensed to Puma by Pfizer for global development and commercialization.
HKI-272 (neratinib) has been described for the treatment of neoplasms [US Patent 6,288,082]. Neratinib is a potent irreversible pan erbB inhibitor. Neratinib is an orally available small molecule that inhibits erbB-1 , erbB-2 and erbB-4 at the intracellular tyrosine kinase domains, a mechanism of action that is different from trastuzumab. Neratinib reduces erbB-1 and erbB-2 autophosphorylation, downstream signaling, and the growth of erbB-1 and erbB-2 dependent cell lines.
Preclinical data suggest that neratinib will have antitumor activity in erbB-1 – and/or erbB 2-expressing carcinoma cell lines, with cellular IC50 <100 nM [Rabindran SK, et al. Antitumor activity of HKI-272, an orally active, irreversible inhibitor of the HER-2 tyrosine kinase. Cancer Research. 2004;64(1 1 ):3958-65].

Neratanib is being developed by Puma Biotechnology. It will be included in the forthcoming I-SPY2 breast cancer trial.[6]
neratinib refers to HKI-272, which has the following core structure:

in its free base form. Optionally, a pharmaceutically acceptable salt or hydrate thereof may be used. The core structure represented above is a particular HKI-272 compound, called HKI-272 or neratinib, which has the chemical name [(2E)-N-[4-[[3-chloro-4- [(pyridin-2-yl)methoxy]phenyl]amino]-3-cyano-7-ethoxyquinolin-6-yl]-4- (dimethylamino)but-2-enamide]. Although currently less preferred, another HKI-272 compound may be used in the place of neratinib. “A HKI-272 compound” refers, in one embodiment, to a compound derived from the core structure of neratinib shown above
The preparation of HKI-272 compounds, of which neratinib is a species, are described in detail in US Patent Application Publication No. 2005/0059678, which is hereby incorporated by reference. See, also, US Patent Nos. 6,288,082, US Patent No. 6,002,008, US Patent No. 6,297,258 and US Patent Application Publication No. 2007/0104721 , which are hereby incorporated by reference. The methods described in these documents can also be used to prepare neratinib and/or the other HKI-272 and substituted 3-quinoline compounds used herein and are hereby incorporated by reference. In addition to the methods described in these documents, International Patent Publication Nos. WO-96/33978 and WO-96/33980, which are hereby incorporated by reference, describe methods that are useful for the preparation of these HKI-272 compounds. Although these methods describe the preparation of certain quinazolines, they are also applicable to the preparation of correspondingly substituted 3- cyanoquinolines and are hereby incorporated by reference.
The term “treating” or “treatment” refers to the administration of the neratinib to a subject to prevent or delay, to alleviate, or to arrest or inhibit development of the symptoms or conditions associated with neoplasms
(E)-N-{4-[3-chloro-4-(2-pyridinylmethoxy)anilino]-3-cyano-7-ethoxy-6-quinolinyl}-4- (dimethylamino)-2-butenamide is an irreversible inhibitor to Her-2 (also known as ErbB-2 or neu) kinase, a member of the epidermal growth factor receptor (EGFR) family. EGFR family members have been implicated in tumorigenesis and associated with poor prognosis in tumor types in humans. The structure of the (E)-N-{4-[3-chloro-4-(2-pyridinylmethoxy)anilino]-3-cyano- 7-ethoxy-6-quinolinyl}-4-(dimethylamino)-2-butenamide in the form of a free base is shown below:

The compound (E)-N-{4-[3-chloro-4 J-(2-pyridinylmethoxy)anilino]-3-cyano-7-ethoxy-6-quinolinyl}- 4-(dimethylamino)-2-butenamide in the form of a free base is described in U.S. Patent No. 6,288,082. The compound is classified, based on the Biopharmaceutical Classification System, as a BCS Class IV compound (low water solubility and low permeability). The free base has low solubility in water, with a water solubility of about 1 μg/ml_ at about pH 7. The water solubility increases with decreasing pH as the compound becomes ionized. This compound is water soluble at gastrointestinal pH, and dissolution is not rate limiting.
Research on Chemical Intermediates, 2012, 09(22),6168
10.1007/s11164-012-0822-4
The Wittig–Horner reaction for the synthesis of neratinib
10.1007/s11164-012-0822-4
The Wittig–Horner reaction for the synthesis of neratinib
…………………
U.S. Patent No. 6,288,082
…………
WO2010048477A2
U.S. Pat. No. 7,126,025 discloses certain novel 4-amino-2-butenoyl chlorides, processes for their preparation and their use as intermediates in the synthesis of pharmaceutically active protein kinase inhibitors, including but not limited to for example HKI-272 and EKB-569.
The sequence illustrated below and summarized in Scheme 1 describes one existing process for preparing HKI-272, (E)-Λ/-(4-(3-chloro-4-(pyridin-2-ylmethoxy)phenylamino)-3- cyano-7-ethoxyquinolin-6-yl)-4-(dimethylamino)but-2-enamide in the form of the maleate salt, also known as Neratinib™.

1 95 eq (COCI)2, cat DMF
O
^
Step 5 OH 16 h HCI

Scheme 1

Scheme 2

Scheme 3. Formation of acid chloride with SOCI2 in DMAc and coupling with a substituted aniline.
SOCl2
/ Nv^-^’C02H HCI DMAc HCI

Scheme 4. Formation of the MW 638 impurity.

Example 4: Process 3
4-Dimethylaminocrotonoyl chloride hydrochloride and its coupling with 6-amino- 4-(3-chloro-4-(pyridin-2-ylmethoxy)phenylamino)-7-ethoxyquinoline-3-carbonitrile (procedure with thionyl chloride and DMAc).
A suspension of 4-dimethylaminocrotonic acid (17.0 g, 97.5 mmol) in DMAc (170 ml_) was cooled to -15 0C under nitrogen atmosphere. Neat thionyl chloride (12.8 g, 7.83 mmol) was added to the slurry at a rate to maintain the temperature in the reactor in the range of -18 to -14 0C (moderate exotherm). The reaction mixture was held at -17 to -15 0C for 4 hrs. A solution of the aminoquinoline (36.2 g, 81.3 mmol) in DMAc (440 ml_) was added to the reactor maintaining the temperature in the -14 to -19 0C range. The resulting mixture was held for 18 hr at approximately -15 0C. At this point HPLC analysis showed residual aniline level at 2.5%. The thick suspension of the hydrochloride salt of the coupled product was quenched with water (200 ml_) maintaining the batch temperature between -5 and -16 0C. The pH of the resulting clear solution was adjusted to 1 1 with a 13% aqueous solution of NaOH (approx. 210 ml_ of the solution was added). The suspension was further diluted with water (350 ml_) and the solids were filtered on a polypropylene cloth filter. The cake was washed with water until neutral pH of the washes and dried first in the nitrogen flow on the filter and then on a tray in vacuum at 45 to 50 0C to afford crude (.=)-/\/-(4-(3-chloro-4-(pyridin-2-ylmethoxy)phenylamino)-3-cyano-7- ethoxyquinolin-6-yl)-4-(dimethylamino)but-2-enamide (42.0 g, 91 %) as a bright-yellow crystalline solid.
………………..
WO2004066919A2
Reaction Scheme Example 1 :
SCHEME 1

(“)

6-(4-N,N-dimethylarninocrotonyt)amido- 4-(4-benzyioxy-3-chloro)arniπo-3-cyano- 7-ethoxyquiπoline, WAY-177820 C31H3[1CIN5θ3 MW 556.07
A suspension of 4-N,N-dimethylaminocrotonic acid hydrochloride in acetonitrile and a catalytic amount of DMF is cooled to 0-10° C. Oxalyl chloride (0.95 eq) is added dropwise and the mixture warmed to 25-30° C and stirred until the chlorinating agent is completely consumed. The light yellow solution is checked for complete consumption of oxalyl chloride by HPLC then cooled to 0-10° C. A cooled solution (0-10° C) of 4-[4-benzyloxy-3-chloro]amino-6-amino-3-cyano-7- ethoxyquinoline in NMP is added dropwise and the mixture is stirred until < 2% of the starting aniline remains. The mixture is added to saturated aqueous sodium bicarbonate, the yellow precipitates are filtered and washed with water. The wet solids are heated to reflux in acetonitrile and clarified hot to remove insolubles. The solution is cooled, the precipitated product filtered and washed with cold acetonitrile. The product is dried (40-50° C, 10 mm Hg, 24 hours) to obtain the final product. Reaction Scheme Example 2:

A solution of 4-N,N-dimethylaminocrotonic acid hydrochloride in tetrahydrofuran (THF) and a catalytic amount of dimethyiformamide (DMF) is cooled to 0-5s C. Oxalyl chloride (0.95 eq) is added dropwise and the mixture warmed to 25-302C and stirred until the chlorinating agent is completely consumed. The orange solution is checked for complete consumption of oxalyl chloride by high- pressure liquid chromatography (HPLC) then cooled to 0-52 C. A solution of 4-[4-(2- pyridylmethoxy)-3-chloro]amino-6-amino-3-cyano-7-ethoxyquinoline is added dropwise and the mixture is stirred until < 0.5% of the starting aniline remains. The reaction is quenched with water and the mixture warmed to 40s C. Aqueous sodium hydroxide is added to bring the pH to 10-11. The resulting precipitates are filtered hot and washed with water. The wet solids are heated to reflux (70-759 C) in acetonitrile:THF (1 :5:1) and the solution cooled slowly to room temperature. The product is filtered and washed with acetonitrile.THF. The product is dried (50e C, 10 mm Hg, 24 hours) to 80-85% yield.
Reaction Scheme Example 3:

4-Dirnethy!amino-but-2-enoic acid |4-(3-chloro-4-fluoro-phenylamino)-3-cvano-7- ethoxy-quinolin-6-vHamide
A. 4-(dimethylamino)-2-butenoyl chloride hydrochloride
A 1 L multi-neck flask equipped with agitator, thermometer, addition funnel, and nitrogen protection is charged with acetonitrile (0.67 kg, 0.85 L) followed by adding dimethylformamide (0.00086 kg, 0.91 mL, d=0.944 g/mL). At ambient temperature, is added 4-dimethylaminocrotonic acid hydrochloride (0.0709 kg) and the mixture stirred until homogeneous. Cool the reaction mixture to (0-10° C) and add oxalyl chloride (0.0473 kg, 0.0325 L, d = 1.45 g/mL) dropwise over (20 minutes) at (0-10° C) followed by a rinse with acetonitrile (0.02 kg, 0.03 L). The temperature (0-10°C) is maintained for about (20 minutes). The temperature of the reaction mixture is adjusted to (22-26° C) over (20 minutes) and maintained over (2 hours). The temperature of reaction mixture is adjusted to (40-45° C) and held for about (5 minutes). Cool the light suspension to about (20-25° C) and check for reaction completion by high-pressure liquid chromatography (HPLC). The reaction is complete when there is < 15 % of the starting material (4-dimethylaminocrotonic acid hydrochloride) present and/or < 2 % of oxalyl chloride (detected as the dimethyl oxalate).
B. 4-Dimethy!amino-but-2-enoic acid |4-(3-chloro-4-fluoro-phenylamino)-3-cyano-7- ethoxy-quinolin-6-yll-amide (crude)
A 3 L multi-neck flask equipped with agitator, thermometer, dip tube, and nitrogen protection is charged N-methyl pyrrolidinone (0.77 kg, 0.75 L, d=1.033 g/mL). At ambient temperature is added 4-[3-chloro-4-fluorophenyl]amino-6-amino-3-cyano-7- ethoxy quinoline (0.0748 kg). The reaction mixture is heated to 40-45° C and maintained for about (15 minutes). The reaction mixture is cooled to (0-10° C) and the light suspension of 4-(dimethylamino)-2-butenoyl chloride hydrochloride in CH3CN added via dip tube and positive nitrogen pressure, over (30-45 minutes) while maintaining the temperature (0-10° C) for at least (2 hours). Reaction completion is monitored by HPLC. The reaction is complete when there is < 2 % of the starting material (4-[3-chloro-4-fluorophenyl]amino-6-amino-3-cyano-7-ethoxy quinoline) present. To a 12 L multi-neck flask equipped with agitator, thermometer, dip tube, and nitrogen protection is charged with water (2.61 kg, 2.61 L) and sodium bicarbonate (0.209 kg) with stirring until a solution is obtained followed by cooling to (20-24° C) to which is transferred the reaction mixture above which contains < 2 % of the starting material (4-[3-chloro-4-fluorophenyl]amino-6-amino-3-cyano-7-ethoxy quinoline), via dip tube and positive nitrogen pressure, to the 12 L flask over about (45-60 minutes) while maintaining the temperature at (20-24° C). The temperature is maintained at (20-24° C) for at least (1 hour). Filter the reaction mixture on a Buchner funnel, rinse with water (3 x 0.40 kg, 3 x 0.40 L), and maintain suction until dripping stops. Dry the product in a vacuum oven at about (50° C) and about (10 mm Hg) for about (28-30 hours). The yield is 78.5 g (86%) at 79.7% strength and 12.3% total impurities.
4-Dimethylamino-but-2-enoic acid r4-(3-chloro-4-fluoro-phenylamino -3-cyano-7- ethoxy-quinolin-6-vn-amide (purified small scale)
First crop: A 6 L multi-neck flask equipped with agitator, condenser, temperature probe, and nitrogen protection is charged with acetonitrile (3.14 kg, 4.00 L) followed by adding 4-dimethylamino-but-2-enoic acid [4-(3-chloro-4-fluoro-phenylamino)-3-cyano-7- ethoxy-quinolin-6-yl]-amide (0.16 kg, 0.167 moles). Heat the mixture to (75-80° C) and hold it for (1 hour). Cool the mixture to (70-75° C) and filter on a pad of diatomaceous earth to remove inorganic salts. Wash the pad with acetonitrile (2 x 0.24 kg, 2x 0.30 L), preheated to (70-75° C). Concentrate the filtrate at (20-30 mm Hg) and a maximum temperature of (40-45° C) to a volume of ( 1.2 L). To the concentrate (slurry) add prefiltered tetrahydrofuran (0.53 kg, 0.60 L). Heat to (65-70° C) to obtain a complete solution. Cool the mixture to (40-45° C) over (0.3 hours). Add seeds and continue cooling to (20-25° C) over (1 hour). Hold at (20-25° C) for a minimum of (18 hours). Collect the solid on a Buchner funnel and wash the collected solid with a prefiltered and precooled at (0-5° C) mixture of acetonitrile/tetrahydrofuran (2/1 by volume) (2 x .06 kg, 2 x 0.08 L). Dry the product in a vacuum oven at (50° C) and (10 mm Hg) for (48 hours) to a loss on drying (LOD) of less than (0.5 %). All washes and concentrates (mother liquors) are saved for further purification.
Second crop:
A 3 L multi-neck flask equipped with agitator, temperature probe, nitrogen protection, and charge with the mother liquors and washes from above. Concentrate by distillation at (20-30 mm Hg) and a maximum temperature of (40-45° C) to a volume of (0.50 L). Collect the solid on a Buchner funnel and wash the solid with prefiltered acetonitrile (0.04 kg, 0.05 L). Dry the solid product in a vacuum oven at (50° C) and (10 mm Hg) for (18 hours). A 1 L multi-neck flask equipped with agitator, condenser, temperature probe, nitrogen protection and charge with prefiltered acetonitrile (0.47 kg, 0.60 L), and the collected solid is heated as a suspension to (70-75° C) over (0.5 hours). Add prefiltered tetrahydrofuran (0.03 kg, 0.03 L) to the suspension while maintaining the temperature at (70-75° C). Cool the solution to (40-45° C) and add seed crystals. Continue cooling to (20-25° C) over (1 hour) and hold for (2 hours). Collect the resulting solid on a Buchner funnel and wash the collected solid with a prefiltered and precooled to (5° C) mixture of acetonitrile/tetrahydrofuran (20/1 by volume) (2 x 0.02 kg, 2 x 0.03 L). Dry the collected solid in a vacuum oven at (50° C) and (10 mm Hg) for (24 hours) to an LOD of less than (0.5 %). The combined yield is 27.5 g + 30.5 g (73%) in 96.2-98.4% strength and 1.5-1.7% total impurities by high pressure liquid chromatography (HPLC).
4-Dimethylamino-but-2-enoic acid f4-(3-chloro-4-fluoro-phenylamino)-3-cvano-7- ethoxy-quinolin-6-vn-amide (purified larger scale)
Acetonitrile, practical (34.0 kg) and 4-dimethylamino-but-2-enoic acid [4-(3- chloro-4-fluoro-phenylamino)-3-cyano-7-ethoxy-quinolin-6-yl]-amide (2.69 kg crude, 1.53 kg at 100% strength) are charged to a purged (100 L) reactor. Acetonitrile, practical (2.0 kg) is used as rinse for funnel and vessel walls. The brown suspension is heated at 70 to 76° C using a jacket temperature not exceeding 85° C, then held at the latter temperature for a minimum of 45 minutes, not exceeding 60 minutes. The resulting suspension is then filtered on the warm-jacketed (70-76° C) 14″ Aurora filter, while maintaining the batch temperature at 70 to 76° C. The filtrates are collected by pump into a purged (100 L) receiver, while keeping their temperature below 50° C. The diatomaceous earth pad is then washed with warm (70 to 76° C) acetonitrile, practical (3 x 2.5 kg). The filtrates and washes in (100 L) receiver are cooled to 20 to 26° C, then transferred into a stainless steel drum. Acetonitrile, practical (2.0 kg) is used as rinse. After cleaning and purging both vessels, the contents of the stainless steel drum is transferred into the (100 L) receiver. Acetonitrile, practical (2.0 kg) is used as a rinse. The batch is heated at 70 to 76° C without exceeding jacket temperature of 85° C. The batch is filtered by pump through a .0 micron single cartridge filter, while maintaining the contents at 70 to 76° C. Warm (70-76° C) acetonitrile, practical (4.0 kg) is used as rinse for vessel, filters, pump and lines. The filtrate and rinse are collected and maintained below 50° C. The batch is adjusted to 10 to 16° C, then concentrated by vacuum distillation to 28 to 33 L volume: expected distillation temperature 20 to 30° C, distillate volume 32 to 37 L. The suspension is heated to 64 to 70° C without exceeding jacket temperature of 85° C. The resulting solution is cooled to 40 to 46° C, then seeded using 4-dimethylamino-but-2~enoic acid [4-(3-chloro-4-fluoro-phenylamino)-3-cyano- 7-ethoxy-quinolin-6-yl]-amide, purified (0.5 g). The mixture is cooled to 20 to 26° C over 1 hour, then held at the latter temperature for a minimum of 2 hours. The suspension is then cooled at -3 to 3° C over 1 hour, then held for a minimum of 1 hour. The solid product is collected on a 16″ Buchner, then washed with cold (0-5° C) acetonitrile-tetrahydrofuran (20-6 v/v) mixture (2 x 2.5 kg). The wet collected solid is recrystallized once more from acetonitrile-tetrahydrofuran (20-6 v/v) to desired purity. The material is dried in a vacuum oven first at 35 to 45° C (target 40° C) for 4 hours, liquid ring pump, then 45 to 55° C (target 50° C) for 4 hours. After high vacuum is applied at the latter temperature, until LOD <0.5% (90° C, 2 hours, full vacuum) and each of acetonitrile, tetrahydrofuran and 1-methyl-2-pyrrolidinone are below 0.2%. The purified drug substance is milled (Comil), then blended. The yield is 1.10 kg (70.1 %, corrected for starting material). The strength of the material is 98.3% and a total impurities of 1.27%.
………………….
N OXIDE
EXAMPLE 19 Formula 57-Compound 19a

19a: (E)-4-((4-((3-Chloro-4-(pyridin-2-ylmethoxy)phenyl)amino)-3-cyano-7-ethoxyquinolin-6-yl)amino)-N,N-dimethyl-4-oxobut-2-en-1-amine oxide
To a solution of compound A (200 mg, 0.36 mmol, 1.0 eq) in CH2Cl2 (20 mL) was added m-CPBA (74 mg, 0.43 mmol, 1.2 eq) and the resulting mixture was stirred at room temperature for 4 h. A saturated aqueous solution of NaHCO3 (20 mL) was then added and the organic layer was separated, dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by preparative TLC (CH2Cl2/MeOH, 10/1, v/v) to give (E)-4-((4-((3-chloro-4-(pyridin-2-ylmethoxy)phenyl)amino)-3-cyano-7-ethoxyquinolin-6-yl)amino)-N,N-dimethyl-4-oxobut-2-en-1-amine oxide (20 mg, 10%) as a yellow solid.
LC-MS (Agilent): Rt 3.03 min; m/z calculated for C30H29ClN6O4 [M+H]+ 573.19. found 573.2.
1H NMR: (400 MHz, CD3OD) δ (ppm): 8.98 (s, 1H), 8.57 (m, 1H), 8.39 (s, 1H), 7.92 (td, J=7.2, 1.6 Hz, 1H), 7.72 (d, J=8.0 Hz, 1H), 7.39 (m, 1H), 7.36 (d, J=2.4 Hz, 1H), 7.28 (s, 1H), 7.24-7.13 (m, 3H), 6.74 (d, J=15.6 Hz, 1H), 5.29 (s, 2H), 4.32 (q, J=6.8 Hz, 2H), 4.20 (d, J=7.2 Hz, 2H), 3.28 (s, 6H), 1.57 (t, J=6.8 Hz, 3H).
……………
Scheme 2 and Scheme 3. Scheme 2

e-Acelamlno^chloro-S-cyano- 7-ethoxy quinoliπe C,4Hi2CIN2O2 +
MW 289.72

25 °C, 5 h 3-Chloro-4-(3-fluorobenzyl)oxy- anillne
C13Hi1CIFNO

MW 251.69
2 h

free base

Scheme 3

6-Acetamlno-4-chloro-3-cyanc~ 7-elhoxy qulnollne C,4H12CIN2O2 +
MW 28972

3-Chlorc-4-fluoronitrobenzene 2-Pyπdyl carblnol 3-Chloro-4-(3-pyndinylmethoxy) 3-Chloro-4-(2-pyrtdlnylmethewy)- C6H3CIFNO2 C6H7NO nitrobenzene anlllne
MW 17555 MW 109 13 C12H9CIN2O3 C12H11CIN2O d=1 1131 g/ml MW 26467 (EM 264) MW 23469
1 h

(HCI salt)
free base
maleate

Example 1
[0078] Synthesis of 3-chloro-4-(2-pyridylmethoxy)nitrobenzene

[0079] 2-pyridinyl carbinol (31.08 g, 1.05 eq) was dissolved in ACN (750 mL) and KOH flakes (85%) were added (20.6 g, 1.25 eq.). The resulting suspension was warmed to 35 °C. A solution of the 3-chloro-4-fluoronitrobenzene (50.0 g, 0.285 mol) in ACN (250 mL) was added at 35-40 °C. The mixture was held for 14 hours. The mixture was then cooled back to 20-25 °C, quenched with H2O (IL) and the resulting slurry filtered and washed with H2O (3 x 100 mL). The resulting product was isolated as a tan solid in 93% yield with a greater than 99.5% purity as determined by HPLC area. Example Ia
[0080] To accomplish the analogous synthesis of 3-chloro-4-(3-fluorobenzyloxy) nitrobenzene, 3-fluorobenzyl alcohol (0.30 kg, 2.39 mole, 1.05 eq) was dissolved in ACN (6.0 L) and to it was added potassium hydroxide flakes (85%) (0.16 kg, 1.25 eq). The resulting suspension was warmed to 35 0C. A solution of the 3-chloro-4-fluoronitrobenzene (0.40 kg, 2.28 mol) in ACN (2.0 L) was added at 35-40 °C. The mixture was held for 18 hours. The mixture was then cooled back to 20-25 °C, quenched with water (8 L) and the resulting slurry filtered and washed with water (2 x 0.40 L). The resulting product was dried at 45 °C, under 10 mm Hg pressure, for 25 hours to give 0.59 kg (92% yield). Example Ib
[0081] To prepare 4-(benzyloxy)3-chloronitrobenzene, benzyl alcohol (0.34 kg, 3.14 mole, 1.10 eq) was dissolved in acetonitrile (1.70 L) and to it was added potassium hydroxide flakes (85%) (0.24 kg, 1.50 eq). The resulting suspension was warmed to 25 0C. A solution of the 3- chloro-4-fluoronitrobenzene (0.50 kg, 2.85 mol, 1.0 eq) in acetonitrile (0.75 L) was added keeping the pot temperature < 45 0C. The mixture was held for 14 h. The mixture was then cooled back to 0-15 0C, quenched with water (2.5 L) and the resulting slurry was filtered and washed with water (2 x 0.50 L). The resulting product was dried at 50 0C, under 10 mm Hg pressure, for 24 hours to give 0.73 kg (97% yield). [0082] Experimental results for the reaction of Example 1 with different bases and solvents are shown in Table 1. The last three entries on Table 1 are large scale runs in which a 5% excess of pyridyl carbinol was used. Table 1 – Preparation of Nitroaryl Intermediate

NA = not applicable
RT = room temperature (20-25 °C)
Example 2
[0083] Preparation of 3-chloro-4-(2-pyridyhnethoxy)aniline from the nitrobenzene product of
Example 1 was accomplished with catalytic hydrogenation using platinum on carbon.

[0084] A typical hydrogenation was done using 6 volumes of THF, 2% by weight of 5%Pt/C (50% water wet), at 25 psi and at 25-30 0C for approximately 4-6 hours. The reaction is slightly exothermic and the temperature will rise to about 30-35 °C. Cooling is necessary to maintain the temperature below 30 0C.
[0085] As a specific example, a mixture of 3-chloro-4-(2-pyridylmethoxy)nitrobenzene (0.15 kg, 0.57 mole) and 2% (w/w) of 5% Pt/C (6.0 g) in tetrahydrofuran (0.90 L) was hydrogenated at 25 psi for at least 5 hours. The mixture was filtered through a celite pad and washed with tetrahydrofuran (0.60 L). The filtrate was distilled to a volume of about 0.75 L and ethanol (1.12 L) was added. Distillation was continued to a volume of about 0.75 L and ethanol (2.85 L) was added. The mixture may be used “as is” in the step of Example 3 below. Example 2 a
[0086] To accomplish an analogous synthesis of 3-chloro-4-(3-fluorobenzyloxy)aniline, zinc (0.464 kg) was added to a mixture of 3-chloro-4-(3-fluorobenzyloxy)nitrobenzene (0.40 kg, 1.42 mole) and ethanol (4.0 L). The mixture was heated to 40-50 °C. A solution of ammonium chloride (0.152 kg) in water (0.80 L) was added over 0.5 hour keeping the pot temperature at 40-50 °C. The mixture was stirred for 2 hours, filtered and washed with hot (40-50 °C) ethanol (2 x 0.40 L). The filtrate was distilled to a volume of about 0.80 L and 2- methyltetrahydrofuran (2.0 L) was added to dissolve the product. Water (0.80 L) and saturated brine (0.40 L) were added and the layers separated. The organic layer was washed with water (0.60 L), and distilled to a volume of about 0.40 L. Ethanol (2.0 L) was added and distillation continued to a volume of 1.2 L. Example 2b
[0087] To prepare 4-(benzyloxy)-3-chloroaniline, a mixture of 4-(benzyloxy)-3- chloronitrobenzene (0.325 kg, 1.23 mole, 1.0 eq) and 1% (w/w) of 5% Pt/C (3.25 g) in isopropanol (3.25 L) was hydrogenated at 25 psi for a minimum of 4.5 h. The mixture was filtered through a celite pad and washed with isopropanol (2.0 L). The filtrates were used as is in the next step.
[0088] Performing the hydrogenation in isopropyl alcohol (PA), methanol (MeOH), or ethanol
(EtOH) may result in the product being contaminated with late eluting impurity that partially precipitates out on standing in solution. It was found that performing the hydrogenation in a solvent where both the product and starting material are soluble, such as tetrahydrofuran
(THF), resulted in greater product purity and required much less solvent. Thus, THF is a preferred solvent for this step. Experimental results showing the effect of different reaction conditions are shown in Table 2. For the larger scale runs, the first aniline intermediate was not isolated (“NI”) before proceeding with the next step.
Table 2 – Hydrogenation to Form First Aniline Intermediate

* Solid impurities noted after reaction completion. ** percent by weight of starting material. Example 3
[0090] Following hydrogenation to form the first aniline intermediate, acid catalyzed coupling was performed to prepare 4~[3-chloro-4-(2-pyridylmethoxy)anilino]-3-cyano-7-ethoxy-6-N- acetylaminoquinoline, as shown below:

[0091] To perform the coupling reaction, the two reactants were heated together in alcohol at 65-78°C over 4-6 hours, yielding the product. The reaction begins as an amber slurry and thickens to a lighter beige slurry as it approaches completion. Upon scaling up from 75 g to 350 g, it proved necessary to add a catalytic amount (0.025 eq.) of methanesulfonic acid to initiate the reaction. As a specific example, 4-chloro-3-cyano-7-ethoxy-6-N- acetylaminoquinoline (0.141 kg, 0.49 mole) was added to the mixture of Example 2, followed by ethanol (0.037 L) to give a suspension. A catalytic amount of methanesulfonic acid (1.17 g) was added at 20-25 C. The resulting slurry was heated to 70-75 C and held for a minimum of 4 hours. Thickening of the slurry was evident after 1.5 hours. Following reaction completion, the mixture was cooled to room temperature and may be used “as is” in the telescoped reaction of Example 4 below. Example 3 a
[0092] To prepare 6-acetamido-4-[3-chloro-4-(3-fluorobenzyloxy)anilino]-3-cyano-7- ethoxyquinoline, ethanol (4.80 L) was added to the aniline solution followed by 4-chloro-3- cyano-7-ethoxy-6-N-acetylaminoquinoline (0.350 kg, 1.11 mole). A catalytic amount of methanesulfonic acid (2.0 ml) was added at 20-250C. The resulting suspension was heated to 70-750C and held for a minimum of 2 h. Thickening of the slurry was evident during this holding period. Following reaction completion, the mixture was used as is in the following telescoped reaction. Example 3 b
[0093] To prepare 6-acetainido-4-[4-(benzyloxy)-3-chloroanilino]-3-cyano-7-ethoxy-quinoline, isopropanol (6.75 L) was added to the aniline solution followed by 4-chloro-3-cyano-7-ethoxy- 6-N-acetylaminoquinoline (0.277 kg, 0.96 mole, 0.78 eq). A catalytic amount of methane sulfonic acid (3.50 ml) was added at 20-250C. The resulting suspension was heated to 80-850C and held for a minimum of 10 hr. Thickening of the slurry was evident during this holding period. Following reaction completion, the mixture was cooled to 25-35 0C, filtered and the cake washed with isopropanol (3 x 0.25 L). The cake was used as is in the following telescoped reaction.
[0094] As solvents EtOH, DMF or other suitable solvent may be used. Experimental results obtained using different solvents and reaction conditions are shown in Table 3. Difficulty filtering the product of this step (noted in several entries on Table 3) was circumvented by not isolating the solid at this point, but telescoping the reaction with the next step. It has been found that on the order of 20 volumes of EtOH were necessary to achieve reasonable stirring, but that the reaction can proceed in only 10 volumes of DMF, without significant loss in purity. [0095] In Table 3, where the entry is labelled NI , the intermediate product was not isolated, but carried into the next reaction step. Table 3 – Coupling Reaction


NR = no reaction, NI = not isolated; ND = not determined; NA = not available
1. Carried through to the deprotection and generation of free base to give 69.5% overall yield.
2. The overall yield after the deprotection and generation of the free base is 76.1%
3. This reaction was not filtered at all but taken as slurry to the next step.
Example 4 – Deprotection
[0096] The deprotection of the quinoline intermediate formed by the coupling reaction using
2N HCl in water is preferred as noted in Table 4 below. As in the previous Examples, the intermediate product of this step is advantageously not isolated, but carried over as a wet cake into the next step.
[0097] Preparation of 4-[3-chloro-4-(2-pyridylmethoxy)anilino]-3-cyano-7-ethoxy-6- aminoquinoline hydrochloride.

[0098] The reaction mixture from the previous step (Example 3) was taken as is and to it was added 2.7N HCl (3.3L) in H2O (16.0 L). The slurry was heated to 700C and held for 19 hours. The resulting slurry was then filtered and rinsed with 1:1 EtOHTH2O (4 x 1.0 L). The product was isolated as a wet cake and carried through to the next step. A small sample was dried at this stage and analyzed. The HCl salt had a strength of 98.9%. Example 4a
[0099] To prepare 6-amino-4-[3-chloro-4-(3-fluorobenzyloxy)anilino]-3-cyano-7- ethoxyquinoline hydrochloride, the reaction mixture from the previous step was taken as is and to it was added ethanol (1.6 L) and concentrated hydrochloric acid (1.38 L) to bring the pH to 1-3. The suspension was held at 70-75 0C for a minimum of 2 h. After 1 h, the mixture thickens and ethanol (0.80 L) was added. After 2 h, water (6.80 L) was added, the mixture stirred for 1 h and then cooled to 35-45 0C and stirred overnight (12 h). The mixture was filtered and rinsed with 1 : 1 ethanol/water (2 x 0.84 L) at 35-45 0C. The product was isolated as a wet cake and carried through to the next step. Example 4b
[00100] To prepare 6-amino-4-[4-(benzyloxy)-3-chloroanilino]-3-cyano-7- ethoxyquinoline hydrochloride, the wet cake from the previous step was taken as is and to it was added a 2 N solution of concentrated hydrochloric acid (1.16 L) in methanol (5.84 L). The suspension was heated to 63-68 0C and held for a minimum of 30 h. The mixture was cooled to 20-300C, filtered and rinsed with methanol (2 x 0.30 L). The product was isolated as a wet cake and carried through to the next step. Table 4 – Deprotection


ND = not determined (the product was used in the next step as a wet cake) NA = not available SM= starting material
Example 5 – Preparation of free base
[0100] The 4-[3-chloro-4-(2-pyridylmethoxy)anilino]-3-cyano-7-ethoxy-6-aminoquinoline HCl salt was converted to the corresponding free base by treatment with 10% potassium carbonate (1.8 L) in MeOH (2.82 L). The mixture was stirred for a minimum of 2.5 hours and the pH was 9-10. The product was filtered, washed with 1:1 methanol/water (3 x 0.19 L) and dried (at 45-50 C at a pressure of 10 mm Hg, for 24 hours) to give 0.186 kg of product with an overall yield of 86% over 4 steps.

Example 5 a
[0101] To prepare 6-amino-4-[3-chloro-4-(3-fluorobenzyloxy)anilino]-3-cyano-7- ethoxyquinoline free base, the 6-amino-4-[3-chloro-4-(3-fluorobenzyloxy)anilino]-3-cyano-7- ethoxyquinoline hydrochloride salt was converted to its corresponding free base by treatment with 10% potassium carbonate (0.22 kg in 2.27 L water) in methanol (7.21 L) until pH was 10. The mixture was stirred for a minimum of 2 h. The beige suspension was filtered, washed with 1:1 methanol/water (2 x 0.84 L) and dried (45-50 0C, 10 mm Hg, 24 h) to give 0.51 kg of product with an overall yield of 99% over 4 steps. Example 5b
[0102] To prepare 6-amino-4-[4-(benzyloxy)-3-chloroanilino]-3-cyano-7-ethoxyquinolme free base, the 6-amino-4-[4-(benzyloxy)-3-chloroamlino]-3-cyano-7-ethoxyqumoline hydrochloride salt was converted to its corresponding free base by treatment with 10% aqueous potassium carbonate (0.213 kg in 2.13 L) in methanol (6.40 L). The mixture was stirred for a minimum of 1.5 h keeping the pH at 9-10. The product was filtered, washed with water (2 x 0.50 L) and dried (50-60 0C, 10 mm Hg, 20 h) to give 0.347 kg of product with an overall yield of 82% over 4 steps.
Example 6 – Side Chain Coupling
[0103] An acid chloride of formula RV(C=O)-Cl, a mixed anhydride or an activated carboxylase R’ 2-(C=O)-LG derived from the corresponding carboxylic acid, may be used to couple a side chain at the 6 position to form a 6-amido-4-amino-3 cyanoquinoline. R’2 may be alkyl of 1-6 carbon atoms, which may be mono- or di-substituted with amino groups or cycloamino groups, or R’2 may be alkenyl of 2-6 carbon atoms which may be mono- or di- substituted with amino groups or cycloamino groups. [0104] Using the 2-step sequence shown below, an activated carboxylate is prepared in situ and coupled with the aniline. Although the acid chloride can be prepared in acetonitile, a better yield was obtained when the acid chloride was prepared in THF. In both cases, the aniline should be dissolved in NMP before amidation. It is believed that formation of product is better due to better solubility of the aniline in a THF/NMP mixture rather than in an ACN/NMP combination.

[0105] The amount of 4-N,N-dimethylaminocrotonic acid needed was 2 equivalents with respect to aniline. A slight undercharge of 1.95 eq of oxalyl chloride was added along with a catalytic amount (3 mol %) of DMF. The acid chloride was formed via the Vilsmeier intermediate. The completion test for the acid chloride reaction consists of quenching an aliquot of the reaction into ethanol and detecting by HPLC the crotonic acid ethyl ester. This method serves as a check to ensure complete consumption of oxalyl chloride. Excess oxalyl chloride will form diethyl oxalate when quenched in ethanol. [0106] The acid chloride is stable after holding for up to 5 hours at 0-10 °C, when decomposition begins. After 20 hours, complete decomposition takes place. If the acid chloride is allowed to warm, decomposition occurs and its effectiveness is diminished. [0107] The quality of the starting crotonic acid also plays a role in this coupling reaction, as commercially available crotonic acid may contain acetic acid. Acetic acid is detrimental to this reaction. 6-N-acetyl quinoline can be formed which is difficult to remove from the final product. The acetic acid can be removed by re-slurrying the crotonic acid in 4 volumes of isopropanol at room tempature, filtering and drying preferably to a level of less than 0.01%. [0108] It was found that the addition of the aniline solution in NMP to the acid chloride gave a better yield as compared to adding the acid chloride to the aniline. The addition is done keeping the temperature at 0-5 °C. The coupling reaction is slow and requires holding overnight at this temperature. It is not desirable to raise the reaction temperature as the stability of the acid chloride diminishes.
[0109] The reaction is quenched using aqueous sodium hydroxide at 40 °C and then filtered at that temperature. Quenching the reaction at 40 0C gives bigger crystals that are easily filterable. It was observed that filtration at 40 °C was faster than at room temperature. The product is recrystallized from a 1.5:1 mixture of acetonitrile:THF (15 volumes) at 70-75 0C. This in-process purification beneficially removes unreacted aniline. The recovery yields are typically greater than 85%.
[0110] To demonstrate a specific synthesis of (E)-N- {4-[3-chloro-4-(2- pyridinylmethoxy)anilino]-3-cyano-7-ethoxy-6-quinolinyl}-4-(dimethylamino)-2-butenamide, a solution of 4-N,N-dimethylaminocrotonic acid hydrochloride (186 g, 1.12 mol) in THF (1.88 L) and a catalytic amount of DMF (2 mL) was cooled to 0-5 °C. Oxalyl chloride (97 mL, 1.09 mol, 0.95 eq) was added dropwise over 45 minutes. The mixture was then warmed to 25-30 °C and stirred for 2 hours. The yellow solution was checked for complete consumption of oxalyl chloride by HPLC, then cooled to 0-5 0C.
[0111] When the reaction is deemed complete, a solution of 4-[4-(2-pyridylniethoxy)-3- chloro]amino-6-amino-3-cyano-7-ethoxyquinoline (250 g, 0.56 mol) in N-methyl-2- pyrolidinone (1.88 L) was added dropwise over 2 hours keeping the temperature at 0-5 °C. The mixture was stirred for at least 3 hours until less than about 2% of the starting aniline remains by HPLC, which takes about 3 hours.
[0112] Upon completion, the reaction was quenched with water (3.0 L), held for 30 minutes and then warmed to 40 °C. Aqueous sodium hydroxide (170 g in 1.25 L water) was added over 1.25 hours to bring the pH to 10-11. The mixture was stirred for an hour, then cooled to room temperature and held for 3 hours. The resulting precipitates were filtered and washed with water (100 mL) and heptane (100 mL). The wet solids were heated to reflux (70-75 °C) in acetonitrile:THF and the solution cooled over 3 hours to room temperature. The product was filtered and washed with cold acetonitrile:THF. The product was dried (40-50 0C, 10 mm Hg, 24 hours) to give 83% uncorrected yield. Example 6a
[0113] In an analogous synthesis of (E)-N- {4-[3-chloro-4-(3-fluorobenzyloxy)anilino]-3- cyano-7-ethoxy-6-qumolmyl}-4-(dimethylamino)-2-butenamide, a solution of 4-N5N- dimethylaminocrotonic acid hydrochloride (108 g, 0.65 mole) in tetrahydrofuran (1.13 L) and a catalytic amount of dimethylformamide (1.2 mL) was cooled to 0-5 °c. Oxalyl chloride (55 mL, 0.62 mole, 0.95 eq) was added dropwise over 50 min. The mixture was then warmed to 25-30 °c and stirred for 2 h then cooled to 0-5 °c. N-methyl-2-pyrrolidinone (0.225 L) was added over 25 min followed by a solution of 6-amino-4-[3-chloro-4-(3- fluorobenzyloxy)]anilino-3-cyano-7-ethoxy-quinoline (150 g, 0.32 mol) in N-methyl-2- pyrrolidinone (1.20 L) added dropwise over 2 hours keeping the temperature 0-5 . The mixture was stirred for at least about 3 hours, warmed to 10-15 °c and stirred for a further 12 hours. The mixture is cooled to 0-10 c, quenched by adding water (1.8 L) over 2 hours, and stirred for 30 minutes. The mixture is warmed to 40 °c. Aqueous sodium hydroxide (101 g in 0.75 L water) was added over 1 hour to bring the pH to 10-11. The mixture was stirred for an hour, filtered warm (40 °c) and washed with water (2 x 0.30 L) until the pH of the last wash was about 7. The wet solids were recrystallized by heating to reflux (70-75 °c) in 60:40 acetonitrile:tetrahydrofuran (2.25 L) and the solution cooled over 3 hours to room temperature. The product was filtered and washed with cold 60:40 acetonitrile:tetrahydrofuran (2 x 0.30 L). The product was dried (40-50 °c, 10 mm Hg, 16 h) to give 0.154 kg (83% yield). Example 6b
[0114] To prepare (E)-N- {4-[4-(benzyloxy)-3-chloroanilino]-3-cyano-7-ethoxy-6-quinolinyl}- 4-(dimethylamino)-2-butenamide free base, a solution of 4-N,N-dimethylaminocrotonic acid hydrochloride (18.6 g, 112 mmole) in acetonitrile (295 ml) and a catalytic amount of dimethylformamide (0.25 mL) was cooled to 0-5 °c. Oxalyl chloride (9.3 mL, 106 mmole, 0.95
Op eq) was added dropwise over 5 min. The mixture was then warmed to 25-30 and stirred for 1-1.5 h then cooled to 0-10 °c. A solution of 6-amino-4-[4-(benzyloxy)-3-cliloroanilino]-3- cyano-7-ethoxy-quinoline (25 g, 56 mmole) in N-methyl-2-pyrrolidinone (175 ml) was added dropwise over 30 min keeping the temperature 0-10 °c. The mixture was stirred for a minimum of 1 h at 0-10 °c. After reaction completion, the mixture was quenched by dropwise addition to a solution of sodium bicarbonate (69.7 g in 870 ml water) over 30 mins. and stirred overnight while warming to room temperature. The mixture was filtered and washed with water (3 x 25 ml). The crude product was recrystallized in refluxing (80-82 °c) acetonitrile (570 ml). The product was dried (45-50 °c, 10 mm Hg, 28 h) to give 12.81 g (41% yield). 1H NMR : δ (DMSO-d6) 9.44 (s, IH, NH), 8.97 (s, IH, Ar), 8.44 (s, IH, Ar), 7.53-7.35 (m, 7H, Ar), 7.35- 7.10 (in, 2H, Ar), 6.78 (dt, IH, -CH2CH=CH-), 6.59 (d, IH, -CH2CH=CH-), 5.21 (s, 2H, OCH2Ph), 4.30 (q, 2H, OCH2CH3), 3.07 (s, 2H, NCH2), 2.18 (s, 6H, N(CHs)2), 1-47 (t, 3H, OCH2CH3).
[0115] Results obtained with different reaction procedures at different degrees of scale-up for synthesis of the 2-pyridylmethoxy analog are shown in Table 5. Table 5 – Side Chain Coupling


* TI = total impurities
[0116] Purificatiuon of the product is conducted by recrystallization in a suitable solvent followed by reslurrying with water followed by additional recrystallization, as necessary. As noted in Table 6, in the synthesis of the 2-pyridylmethoxy analog, several trials in different solvents did not result in the isolation of a single polymorphic form of the product. Table 6


Example 7 – Formation of Salt
[0117] The free base is hygroscopic and undergoes hydrolysis readily. Forming a salt of the compound, such as a fumarate or mesylate salt, stabilizes the molecule and renders the compound more soluble. The most preferred salt is a maleate salt, which has been found to be highly crystalline and to exist substantially as a single polymorph as shown by DSC thermogram in Fig. 1.
[0118] Recrystallizing the product in the presence of an acid has been found to yield a stable salt form of the product. Experimental results achieved utilizing different solvents for the recrystallization are set forth in Table 7. As seen in Table 7, an improvement is observed when n-propanol/water is used as the solvent system. A maleate salt is the most preferred, as it exists in a single polymorphic form. Table 7 – Recrystallization



Preparation of (E)-N- {4-[3-chloro-4-(2-pyridinylmethoxy)anilino]-3-cyano-7-ethoxy-6- quinolinyl} -4-(dimethylamino)-2-butenamide maleate, WAY- 179272-B
[0120] (E)-N-{4-[3-chloro-4-(2-pyridinylmethoxy)anilino]-3-cyano-7-ethoxy-6-quinolinyl}-4- dimethylamino)-2-butenamide crude free base (0.1 kg, 0.159 mole) and maleic acid (0.019 kg, 0.164 mole) were dissolved at 40-50 in a 10% water/n-propanol mixture (1.20 L). The hot solution was clarified and cooled over 2 h to room temperature and held for 12-15 hr. The product was filtered and washed with 10% water/n-propanol (2 x 0.15 L). The product was dried (50 °c, 10 mm Hg, 24 h) to give 94.4 g (88% yield). DSC: 204 °c (single crystal form). 1H NMR : δ (DMSO-d6) 9.73 (s, IH, NH), 9.62 (s, IH, NH), 8.93 (s, IH, Ar), 8.60 (dd, IH, Ar), 8.50 (s, IH, Ar), 7.88 (dd, IH, Ar), 7.58 (d, IH, Ar), 7.40 (m, 3H, Ar), 7.24 (m, 2H, Ar), 6.75 (d, 2H, -CH=CH-), 6.03 (s, 2H, HOOC-CH=CH-COOH), 5.29 (s, 2H, OCH2PVr), 4.33 (q, 2H, OCH2CH3), 3.89 (s, 2H, NCH2), 2.76 (s, 6H, N(CH3)2), 1.47 (t, 3H, OCH2CH3). 13C NMR : δ (DMSO-d6) 168.0, 163.2, 156.9, 154.2, 153.2, 151.9, 151.3, 149.8, 148.5, 137.8, 136.5, 134.7, 133.4, 132.2, 128.0, 126.6, 124.9, 123.8, 122.3, 122.2, 117.9, 116.4, 115.1, 113.9, 109.5, 88.1, 72.0, 65.3, 57.8, 43.1, 14.9.
Example 7a
To prepare (E)-N- {4-[3-chloro-4-(3-fluorobenzyloxy)anilino]-3-cyano-7-ethoxy-6- quinolinyl}-4-(dimethylamino)-2-butenamide dimaleate,
(E)-N- {4-[3-chloro-4-(3- fluorobenzyloxy)anilino]-3-cyano-7-ethoxy-6-quinolinyl}-4-dimethylamino)-2-butenamide crude free base (0.516 kg, 0.90 mole) and maleic acid (0.214 kg, 1.84 mole) were dissolved at 40-50 °c in a 6.5% water/n-propanol mixture (12.60 L). The hot solution was clarified, rinsed with 5% water/n-propanol (0.52 L) and n-propanol (2.0 L). The mixture was held at 45 for 3 hr, cooled over 2 h to room temperature and held overnight. The mixture was further cooled to 5-10 °c. The product was filtered and washed with cold 5% water/n-propanol (0.52 L). The product was dried (45 °c, 10 mm Hg, 16-24 h) to give 0.586 kg (81% yield). DSC: 184 °c (single crystal form). 1HNMR : δ (DMSO-d6) 9.77 (s, IH, NH), 8.95 (s, IH, Ar), 8.53 (s, IH, Ar), 7.49-7.16 (m, 8H, Ar), 6.78 (m, 2H, -CH=CH-), 6.15 (s, 4H, 2 x HOOC-CH=CH-COOH), 5.26 (s, 2H, OCH2PyT), 4.33 (q, 2H, OCH2CH3), 3.97 (dd, 2H, NCH2), 2.82 (s, 6H, N(CEb)2), 1.47 (t, 3H, OCH2CH3). 13C NMR : δ (DMS0-d6) 167.0, 163.8, 162.3, 160.6, 153.6, 152.2, 151.3, 150.8, 139.5, 139.4, 133.7, 133.2, 132.2, 131.8, 130.5, 130.4, 127.4, 126.1, 124.3, 123.3, 121.7, 116.9, 115.7, 114.8, 114.5, 114.4, 114.1, 113.8, 113.1, 108.1, 87.2, 69.5, 64.6, 56.9, 42.1, 14.2. Example 7b
[0122] To prepare (E)-N- {4-[4-(benzyloxy)-3-chloroanilino]-3-cyano-7-ethoxy-6-quinolinyl}- 4-(dimethylamino)-2-butenamide maleate, (E)-N- {4-[4-(benzyloxy)-3-chloroanilino]-3-cyano- 7-ethoxy-6-quinolinyl}-4-dimethylamino)-2-butenamide crude free base (2.0 g, 3.6 mmole) and maleic acid (0.43 g, 3.7 mmole) were mixed at 40-50 c in a 10% water/n-propanol mixture (24 ml) for 2 hr. The mixture was cooled to ambient temperature, filtered and washed with 10% water/n-propanol (2 x 3 ml). The product was dried (40 °c, 10 mm Hg, 24 h) to give 0.32 g (13% yield). 1HNMR : δ (DMSO-d6) 9.75 (s, IH, NH), 8.95 (s, IH, Ar), 8.49 (s, IH, Ar), 7.49-7.37 (m, 7H, Ar), 7.23 (dd, 2H, Ar), 6.78 (s, 2H, -CH2CH=CH-), 6.06 (s, 2H, HOOC- CH=CH-COOH), 5.22 (s, 2H, OCH2Ph), 4.31 (q, 2H, OCH2CH3), 3.93 (s, 2H, NCH2), 2.79 (s, 6H, N(CH3)2), 1.46 (t, 3H, OCH2CH3).13C NMR : δ (DMSO-d6) 167.9, 163.1, 154.2, 153.3, 152.1, 151.3, 148.5, 137.3, 136.3, 134.5, 133.2, 132.3, 129.3, 129.2, 128.7, 128.3, 128.2, 128.0, 126.7, 124.9, 122.4, 117.9, 116.4, 115.2, 113.9, 109.5, 88.0, 71.1, 65.3, 57.7, 43.0, 15.0. [0123] (E)-N-{4-[4-(benzyloxy)-3-chloroanilino]-3-cyano-7-ethoxy-6-quinolinyl}-4- dimethylamino)-2-butenamide crude free base (2.0 g, 3.6 mmole) and maleic acid (0.43 g, 3.7 mmole) were mixed at 40-50 °c in a 10% water/n-propanol mixture (24 ml) for 2 hr. The mixture was cooled to ambient temperature, filtered and washed with 10% water/n-propanol (2 x 3 ml). The product was dried (40 °c, 10 mm Hg, 24 h) to give 0.32 g (13% yield). 1H NMR : δ (DMSO-d6) 9.75 (s, IH, NH), 8.95 (s, IH, Ar), 8.49 (s, IH, Ar), 7.49-7.37 (m, 7H, Ar), 7.23 (dd, 2H, Ar), 6.78 (s, 2H, -CH2CH=CH-), 6.06 (s, 2H, HOOC-CH=CH-COOH), 5.22 (s, 2H, OCH2Ph), 4.31 (q, 2H, OCH2CH3), 3.93 (s, 2H, NCH2), 2.79 (s, 6H, N(CH3)2), 1.46 (t, 3H, OCH2CH3). 13C NMR : δ (DMSO-d6) 167.9, 163.1, 154.2, 153.3, 152.1, 151.3, 148.5, 137.3, 136.3, 134.5, 133.2, 132.3, 129.3, 129.2, 128.7, 128.3, 128.2, 128.0, 126.7, 124.9, 122.4, 117.9,
116.4, 115.2, 113.9, 109.5, 88.0, 71.1, 65.3, 57.7, 43.0, 15.0.
……………….
TABLE 1 1. STRUCTURES OF DEGRADATION PRODUCT AND PROCESS IMPURITIES

N-{4-[3-chloro-4-(2- (E)-4-({4-[3-chloro-4-(2- N -{4-[3-chloro-4-(2- pyrιdιnylmethoxy)anιlιno]-3-cyano-7- pyrιdιnylmethoxy)anιlιno]-3-cyano-7- pyrιdιnylmethoxy)anιlιno]-3-cyano-7-ethoxy- ethoxy-6-quιnolιnyl}acetamιde ethoxy-6-quιnolιnyl}amιno)-N,N,N- 6-quιnolιnyl}-N2,N2-dιmethylethanedιamιde trιmethyl-4-oxo-2-buten-1-amιnιum
Exact Mass 487 14 Exact Mass 544 16
Exact Mass 571 22
Process Impurity I Process Impurity J

SCHEME 1

The reaction of the free base and maleic acid occurs at an elevated temperature of from about 40 0C to about 60 0C, preferably between about 4O0C to about 5O0C. The ratio of watenn- propanol may vary, for example between about 1 :10 to about 1 :5, and the optimal ratio of watenn-propanol is about 1 :9. The water-alcohol solution may comprise from about 5% to about 20% by volume water and from about 80% to about 95% by volume alcohol. The alcohol may be n-propanol. In one embodiment, the water-alcohol solution comprises about 10% by volume water and about 90% by volume n-propanol. The volume of the solvent solution may be between about 8 to about 25 volumes, including about 10 to about 12 volumes. About 1.0-1.2 equivalents of maleic acid is used per equivalent of the free base, preferably about 1.03 equivalents of maleic acid per equivalent of the free base.
The resulting solution of the maleate salt may be clarified by filtration prior to cooling. The cooling step may be continued until the solution reaches a temperature of about 45°C or less, including a temperature of about 39°C or less, and more preferably to about 300C or less. In one embodiment, the solution is filtered after cooling to about room temperature, preferably from about 230C to about 25 0C. Typically, the maleate salt begins to crystallize out of solution once the temperature reaches 370C or below. The solution may be allowed to sit for at least 12 hours, preferably about 12 to about 15 hours at room temperature, and is then filtered and washed to recover the crystalline maleate salt product. The resulting filter cake may be washed with the same or a different water-alcohol solution to obtain the product. The product may be dried to obtain crystalline (E)-N-{4-[3-chloro-4-(2-pyridinylmethoxy)anilino]-3-cyano-7- ethoxy-6-quinolinyl}-4-(dimethylamino)-2-butenamide maleate. At this point, the maleate salt product recovered and isolated is typically in the form of the monohydrate form of the maleate salt.
……………
The present invention relates to a process for preparing that imatinib (neratinib, HKI-272) is a new method for its preparation and its intermediates in the preparation to the application that imatinib


[0155] Example 14 (E)-N-(4 – (3 – chloro-4 – (2 – pyridyl) phenyl) amino] _3_ ethoxy-quinolin-6-cyano-_7_ – yl) -4 – dimethylamino-2 – butene amide
[0156]

[0157] Compound of Example 13 (20mg, 0. 037mmol) was dissolved in DMF was added potassium carbonate (10mg, 0. 07mmol), dimethylamine hydrochloride (5mg, 0. 06mmol), at room temperature for I hour, after , the reaction mixture was dropped into water, stirred for 10 minutes, filtered, washed with water and dried to give the title compound 1511 ^ 75% yield.1HNMR (300MHz, DMS0_d6): δ I. 5 (t, 3H, J = 6 · 8,13. 8), 2. 2 (br s, 6H), 3. I (d, 2H, J = 3. 8 ), 4. 3 (q, 2H, J = 7. 0,14. 2), 5. 2 (s, 2H),
6. 6 (d, 1H, J = 15. 0), 6. 8 (m, 1H), 7. 1-7. 3 (m, 2H), 7. 3-7. 4 (m, 3H), 7. 6 (d, 1H, J = 3. 9),
7. 9 (d, 1H, J = 3. 9), 8. 5 (s, 1H), 8. 6 (d, 1H, J = 3. 9), 9. 0 (s, 1H), 9. 5 (s, 1H), 9. 6 (s, 1H). ESI-MS: [M + H] + = 557. 3.
GOING BACKWARDS…………………
[0152] Example 13 (E) -4 – bromo-N-(4 – (3 – chloro-4 – (2 – pyridyl) phenyl) amino] _3_ cyano _7_ ethoxyquin -6 – yl) -2 – butene amide
[0153]

[0154] Example 12 Compound (100mg, 0. 2mmol) was suspended in carbon tetrachloride was added NBS (40mg,
O. 22mmol), benzoyl peroxide (2mg, 0. Olmmol), nitrogen, refluxed for 10 hours, the reaction solution directly mixed baby gel, silica gel column chromatography to obtain the title compound isolated 60mg, yield 51%. 1HnmrgoomHz, cdci3): δ i.6 (t, 3H, J = 6. 8,13. 7), 2. 0 (d, 2H, J = 6. 9), 4. 3 (q, 2H, J = 7. 2,13. 8), 5. 3 (s, 2H), 6. I (d, 1H, J =
15. 0), 7. 0 (m, 1H), 7. 2 (m, 1H), 7. 3 (s, 1H), 7. 4 (s, 1H), 7. 6 (d, 1H, J = 8. 2), 7. 8 (d, 1H, J =
7. 6), 8. 0 (s, 1H), 8. 5 (s, 1H), 8. 6 (d, 1H, J = 4. 7), 9. 2 (s, 1H). ESI-MS: [M + H] + = 594. I.
……………
Optimization of 6,7-disubstituted-4-(arylamino)quinoline-3-carbonitriles as orally active, irreversible inhibitors of human epidermal growth factor receptor-2 kinase activity
J Med Chem 2005, 48(4): 1107
J Med Chem 2005, 48(4): 1107
(E)-N-{4-[3-Chloro-4-(2-pyridinylmethoxy)anilino]-3-cyano-7-ethoxy-6-quinolinyl}-4-(dimethylamino)-2-butenamide (25o).
This compound was prepared as a yellow solid (0.86 g, 85%) by the method described for 25g using 0.65 g (1.81 mmol) of 23 and 0.42 g (3.62 mmol) of 3-chloro-4-(2-pyridinylmethoxy)aniline:
HRMS (ES+) m/z 557.205 89 (M + H)+1, Δ = −0.36 mmu;
1H NMR (DMSO-d6) δ 9.62 (s, 1H), 9.49 (s, 1H), 8.96 (s, 1H),
8.60 (d, 1H, J = 3.9 Hz), 8.47 (s, 1H),
7.88 (t, 1H, J = 3.9 Hz), 7.58 (d, 1H, J = 3.9 Hz),
7.39−7.35 (m, 3H), 7.26 (d, 1H, J = 7.8 Hz),
7.19 (d, 1H, J = 8.1 Hz), 6.81−6.73 (m, 1H),
6.59 (d, 1H, J = 7.8 Hz), 5.28 (s, 2H),
4.30 (q, 2H, J = 6.9 Hz),
3.07 (d, 2H, J = 3.9 Hz),
2.17 (s, 6H),
1.46 (t, 3H, J = 3.9 Hz).
Anal. (C30H29ClN6O3·1.1H2O) C, H, N.
INTERPRETATION
1H NMR : δ (DMSO-d6)
9.44 (s, IH, NH),
8.97 (s, IH, Ar),
8.44 (s, IH, Ar),
7.53-7.35 (m, 7H, Ar),
7.35- 7.10 (in, 2H, Ar),
6.78 (dt, IH, -CH2CH=CH-),
6.59 (d, IH, -CH2CH=CH-),
5.21 (s, 2H, OCH2Ph),
4.30 (q, 2H, OCH2CH3),
3.07 (s, 2H, NCH2),
2.18 (s, 6H, N(CHs)2),
1-47 (t, 3H, OCH2CH3).
References
- “Definition of neratinib – National Cancer Institute Drug Dictionary”. Retrieved 2008-12-01.
- Rabindran SK, Discafani CM, Rosfjord EC, et al. (June 2004). “Antitumor activity of HKI-272, an orally active, irreversible inhibitor of the HER-2 tyrosine kinase”. Cancer Res. 64 (11): 3958–65. doi:10.1158/0008-5472.CAN-03-2868. PMID 15173008.
- ClinicalTrials.gov NCT00398567 A Phase 1/2 Study Of HKI-272 In Combination With Herceptin In Subjects With Advanced Breast Cancer
- “Puma Acquires Global Rights to Pfizer’s Phase III Breast Cancer Drug Neratinib”.
- Minami Y, Shimamura T, Shah K, et al. (July 2007). “The major lung cancer-derived mutants of ERBB2 are oncogenic and are associated with sensitivity to the irreversible EGFR/ERBB2 inhibitor HKI-272″. Oncogene 26 (34): 5023–7. doi:10.1038/sj.onc.1210292.PMID 17311002.
- http://www.reuters.com/article/idUSN1612347120100317 “Breast cancer study aims to speed drugs, cooperation” March 2010
- Sequist L.V., Besse B., Lynch T.J. and all; Neratinib, an Irreversible Pan-ErbB Receptor Tyrosine Kinase Inhibitor: Results of a Phase II Trial in Patients With Advanced Non-Small-Cell Lung Cancer., J. Clin. Oncol., 2010, May 17.
PubMed PMID: 20479403. - Belani CP. The role of irreversible EGFR inhibitors in the treatment of non-small cell lung cancer: overcoming resistance to reversible EGFR inhibitors. Review. Cancer Invest. 2010, 28(4), 413-423. Review.
PubMed PMID: 20307200. - TSOU H-R ET AL: “Optimization of 6,7-Disubstituted-4-(arylamino)quinoline-3 -carbonitr iles as Orally Active, Irreverible Inhibitors of HEGFR-2 Kinase Activity” JOURNAL OF MEDICINAL CHEMISTRY, AMERICAN CHEMICAL SOCIETY, WASHINGTON, US, vol. 48, 27 January 2005 (2005-01-27), pages 1107-1131, XP002414228 ISSN: 0022-2623 cited in the application
- Optimization of 6,7-disubstituted-4-(arylamino)quinoline-3-carbonitriles as orally active, irreversible inhibitors of human epidermal growth factor receptor-2 kinase activity
J Med Chem 2005, 48(4): 1107
2-20-2009
|
Novel Combinational Use of Sulfonamide Compound
| |
9-12-2008
|
Multi-Functional Small Molecules as Anti-Proliferative Agents
| |
5-11-2007
|
Antineoplastic combinations with mTOR inhibitor,herceptin, and/or hki-272
| |
11-31-2006
|
Methods of synthesizing substituted 3-cyanoquinolines and intermediates thereof
| |
11-31-2006
|
Methods of synthesizing 6-alkylaminoquinoline derivatives
| |
10-25-2006
|
Synthesis of 4-(amino)-2-butenoyl chlorides and their use in the preparation of 3-cyano quinolines
|
5-30-2012
|
Amide derivative for inhibiting the growth of cancer cells
| |
9-21-2011
|
Maleate salts of (E)-N-{4-[3-chloro-4-(2-pyridinylmethoxy)anilino]-3-cyano-7-ethoxy-6-quinolinyl}-4-(dimethylamino)-2-butenamide and crystalline forms thereof
| |
8-26-2011
|
COMPOUNDS THAT MODULATE EGFR ACTIVITY AND METHODS FOR TREATING OR PREVENTING CONDITIONS THEREWITH
| |
5-7-2010
|
Antineoplastic Combinations of 4-Anilino-3-Cyanoquinolines and Capecitabine
| |
4-30-2010
|
METHOD FOR PREDICTION OF THE EFFICACY OF VASCULARIZATION INHIBITOR
| |
4-16-2010
|
METHOD FOR ASSAY ON THE EFFECT OF VASCULARIZATION INHIBITOR
| |
3-19-2010
|
PHARMACEUTICAL COMPOSITIONS OF AN SRC KINASE INHIBITOR AND AN AROMATASE INHIBITOR
| |
2-26-2010
|
Heterocyclic N-Oxides as Hypoxic Selective Protein Kinase Inhibitors
| |
12-18-2009
|
Antineoplastic Combinations Containing HKI-272 and Vinorelbine
| |
12-4-2009
|
ANTINEOPLASTIC COMBINATIONS WITH mTOR INHIBITOR, TRASTUZUMAB, AND/OR HKI-272
|
8
ERLOTINIB






ERLOTINIB
 | |
|---|---|
 | |
Erlotinib
N-(3-ethynylphenyl)-6,7-bis(2-methoxyethoxy)
quinazolin-4-amine
quinazolin-4-amine
17 jan 2013
The USFDA has accepted for filing Astellas Pharma’s supplemental new drug application (sNDA) for Tarceva (erlotinib) tablets for a genetically distinct form of advanced lung cancer.
Astellas is seeking approval to use Tarceva as the first-line therapy to treat patients with EGFR activating mutation-positive locally advanced or metastatic non-small cell lung cancer (NSCLC).
The sNDA, which was granted priority review status, included data from EURTAC trial, a randomized, controlled Phase 3 study designed to assess the use of Tarceva compared to platinum-based chemotherapy in NSCLC patients with EGFR activating mutations.
Astellas Pharma Global Development medical oncology head, vice president Stephen Eck said the FDA granted an expedited six-month review of the application.
“We are proud of Tarceva’s already approved indications for the maintenance and relapsed advanced NSCLC settings,” Eck added.
“If approved, people with a genetically distinct form of lung cancer could have a potential new personalized medicine for use as a first-line treatment.”
The cobas EGFR Mutation Test, developed by Roche Molecular Diagnostics, is a companion diagnostic for which a pre-market approval application was also submitted to the regulatory body.
Erlotinib hydrochloride (trade name Tarceva) is a drug used to treat non-small celllung cancer, pancreatic cancer and several other types of cancer. It is a reversibletyrosine kinase inhibitor, which acts on the http://en.wikipedia.org/wiki/Epidermal_growth_factor_receptor” rel=”nofollow”>epidermal growth factor receptor (EGFR). It is marketed in the United States by Genentech and OSI Pharmaceuticals and elsewhere by Roche
Erlotinib is an EGFR inhibitor. The drug follows Iressa (gefitinib), which was the first drug of this type. Erlotinib specifically targets the epidermal growth factor receptor (EGFR)tyrosine kinase, which is highly expressed and occasionally mutated in various forms of cancer. It binds in a reversible fashion to the adenosine triphosphate (ATP) binding site of the receptor.[1] For the signal to be transmitted, two EGFR molecules need to come together to form a homodimer. These then use the molecule of ATP to trans-phosphorylate each other on tyrosine residues, which generates phosphotyrosine residues, recruiting the phosphotyrosine-binding proteins to EGFR to assemble protein complexes that transduce signal cascades to the nucleus or activate other cellular biochemical processes. By inhibiting the ATP, formation of phosphotyrosine residues in EGFR is not possible and the signal cascades are not initiated.
Erlotinib hydrochloride (1), chemically named as N-(3-ethynylphenyl)-6,7-bis-(2-meth- oxyethoxy)-4-qumazolimmine monohydro chloride, is an inhibitor of oncogenic and proto- oncogenic protein tyrosine kinases, e.g. epidermal growth factor receptor (EGFR). Erlotinib is therefore useful in the treatment of proliferative disorders and is currently marketed for the treatment of lung cancer and pancreatic cancer.
(Erlotinib Hydrochloride)
(1)
It has been reported that erlotinib hydrochloride can exist in different polymorphic forms. The manufacturing process for many pharmaceuticals is hindered by the fact that the organic compound which is the active ingredient can exist in more than one polymorphic form. It is essential in pharmaceutical development to ensure that the manufacturing process for the preparation of the active ingredient affords a single polymorph with a consistent level of polymorphic purity. If the manufacturing process produces a product with varying degrees of polymorphic purity and/ or or where the process does not control polymorphic inter-conversion, it could lead to serious problems in dissolution and/ or bioavailability in the finished pharmaceutical composition comprising the active ingredient, Erlotinibhydrochloride is disclosed in patent US 5,747,498 and details of the disclosed method for the preparation of erlotinib hydrochloride are described in Scheme 1.
Scheme 1
4-Chloro-6,7-bis-(2-methoxyed oxy)qiunazoline (2) was reacted with 3-emynylaniline (3) or its hydrochloride salt using various solvents and pyridine as a base to yield erlotinib hydrochloride (1) which was treated widi a biphasic mixture consisting of saturated aqueous NaHC03, chloroform and methanol, to formerlotinib base (4). The base (4) obtained in the organic phase was purified by flash chromatography to afford purified erlotinib base. The purified base was further treated with hydrochloric acid in the presence of diethyl ether and chloroform to yield erlotinib hydrochloride. This isolation of purified erlotinib base required the use of a lengthy workup process including column chromatography and required the chlorinated solvent, chloroform, which is not particularly suitable £01 commercial production of pharmaceuticals. Furthermore, the p irification by column chromatography is neither economical nor feasible at industrial scale. In addition, substantially pure erlotinib could not be obtained. Two crystalline forms of erlotinib hydrochloride (polymorph A and polymorph B), were characterized by XRPD in patent application, WO 01/34574. Erlotinib hydrochloride can be obtained in form A or in a mixture of polymorph A and B, by refluxing 3-ethynylaniline and 4-chloro-6,7-bis-(2-methoxyemoxy)-qitiiiazoline in a mixture of toluene and acetonitrile. This afforded polymorph A or a mixture of polymorph A and B. It was also disclosed that the formation of polymorph A was favoixred by reducing the amounts of acetonitrile with respect to toluene. Furthermore, erlotinibhydrochloride polymorph A can be converted into polymorph B by refluxing the polymorph A with alcohol/water. Consequently, in the disclosed methods, there was always contamination of form A with form B and vice-versa. In addition, the products of the reaction are not chemically pure and difficult to purify thereafter. Consequently, these methods are not suitable for preparation of commercial quantities of pure polymorph A.
A process for the preparation of erlotinib hydrochloride, polymorph E by condensation reaction of 3-emynylaiiiline and 4-chloro-6,7-bis-(2-memoxyethoxy)quii azoline in ( , , )- trifiuorotoluene and HC1 was disclosed in U.S. Patent application 2004/0162300. Polymorph E was characterized by XRPD, IR and melting point. However, (α,α,α)- trifluorotoluene is a highly flammable and dangerous solvent for the environment and is not suitable for commercial production. A process for the preparation of erlotinib hydrochloride, polymorph A by reaction of erlotinib base widi aqueous or gaseous HC1 was disclosed in US 2009/0131665. In this method, toluene, a mixture of toluene and methanol, TBME, ethyl acetate, 1-butanol or MIBK were used as a solvent. However, when DCM, diethyl ether, isopropyl acetate, was used as a solvent, polymorph B was formed. In practice, it has been found that the disclosed methods are inconsistent and afford polymorphic mixtures. In particular, example 1 of US 2009/131665 was repeated and erlotinib hydrochloride was obtained with only 97% purity. In addition, XRPD analysis showed d at the example afforded form B or mixtures of forms A and B. Furthermore, several crystallizations of erlotinib hydrochloride, obtained from repetition of the example, using various solvents and their combinations would not yield a product pure enough to comply with ICH guidelines.
A process for the preparation of a hydrate of erlotinib hydrochloride comprising crystallization of erlotinib hydrochloride using water as solvent, preferably in the absence of organic solvent was disclosed in US 20080167327. This patent also disclosed the process to prepare hemihydrate polymorph form I as well as form II.
A process for the preparation of erlotinib hydrochloride, polymorph M, N and P by reaction of erlotinib base and aqueous or gaseous HC1 dissolved in organic solvents was disclosed in WO 2008/102369.
A process for the preparation of erlotinib hydrochloride by condensation reaction of 4- chloro~6,7-bis-(2-me oxyemoxy)-quinazoline and 3-ethynylaniline in isopropyl alcohol as a solvent and pyridine as a base was disclosed in Molecules Journal (Vol, 11, 286, 2006) but no details on the polymorph were disclosed.
A method for the preparation of erlotinib hydrochloride polymorph A comprising passing hydrochloride gas onto solid erlotinib base containing residual amounts of isopropanol was disclosed in WO 2010/040212. However, in practice it was found that the process did not afford chemically or polymorphically pure product. Repetition of example 1 (page 8) of WO 2010/040212 to prepare erlotinibhydrochloride, by reaction of erlotinib base and gaseous HQ in IPA as a solvent, afforded a mixture of polymorph A and polymorph B (as checked by XRPD).
A process for the preparation of acid salts of erlotinib by reaction of 4-chloro-6,7-bis-(2- memoxyemoxy)-quinazoline and 3-emynykniline or an acid salt of 3-emynylaniline under acidic conditions to form the corresponding erlotinib salt was disclosed in US 2010/0094004. In order to complete the reaction, several hours (6 hours) of reflux was required and hence it is not a cost effective process. In addition, in practice it was found that the process did not afford chemically or polymorplxLcally pure product. A process £oi the preparation of erlotinib base, polymorph Gl, G2 and G3 was disclosed in WO 2009/002538 and WO 2010/05924.
Scheme 2
A method for the preparation of eiiotinib hydrochloride was disclosed in US 2009/0306377. The method, illustrated in Scheme 2, involves treating 6,7-dimethoxy- 4(3H)-quinazolone (5) with hydrobiOmic acid or pyridine-hydrochloric acid to afford 6,7- dihydroxy-4(3H)-quinazolone (6), which was diacetylated with acetic anhydride to afford diester (7), which was treated with oxalyl chloride/DMF to afford 4-chloro-6,7- ctiacetoxyquinazoline (8). Compound (8) was condensed with 3-e ynylaniline to afford JV- (3-ethynylphenyl)-6,7-dihydfoxy-4-quinazolinamine hydrochloride (9), which was converted into the diol N-(3-emynylphenyl)-6,7-dmyckOxy-4-quinazolinamine (10) by treatment with aqueous ammonia/methanol. The diol (10) was treated with 2-iodo-ethylmethyl ether to yield compound (4) which on treatment with HC1 afforded erlotinib hydrochloride (1). However, this preparation of erlotinib hydrochloride is a long synthetic route and gives low yields and requires very toxic reagents like pyridine, HBi and controlled reagents like acetic anhydride. Hence, it is not suitable for large scale production. Object of the invention
The priot art processes described above for the preparation of erlotinib and its salts have major disadvantages with respect to the formation and removal of process related chemical and polymorphic impurities; poor commercial viability due to die use of hazardous reactants; expensive, time consuming separation methods such as column chromatography and/ or low yields and purity of final and intermediate products.
As the commercial production of erlotinib hydrochloride is of great importance, for the treatment of cancer, and in view of the above disadvantages associated with the prior art there is a real need for alternative and improved processes for the preparation of erlotinib hydrochloride which do not involve multiple steps and further eliminates the need for cumbersome purification techniques, particularly for the removal of the chemical and polymorphic impurities. The alternative processes must be economical and high yielding and provide erlotinib and its salts with a high degree of chemical and polymorphic purity.
U.S. Patent No. 5,747,498 disclosed 4-(substituted phenylamino) quinazoline derivatives, processes for their preparation, pharmaceutical compositions in which they are present and method of use thereof. These compounds are Tyrosine Kinase Inhibitors and are useful in the treatment of hyperproliferative diseases, such as cancers, in mammals. Among them, erlotinib hydrochloride, chemically N-(3-ethynylphenyl)-6,7-bis(2-methoxy ethoxy)-4-quinazolinamine hydrochloride is a selective inhibitor of the erbB family of oncogenic and protooncogenic protein tyrosine kinases, such as epidermal growth factor receptor (EGFR), and is useful for the treatment of proliferative disorders, such as cancers, particularly non small cell lung cancer, pancreatic cancer, ovarian cancer, breast cancer, glioma, head cancer or neck cancer.
Polymorphism is defined as “the ability of a substance to exist as two or more crystalline phases that have different arrangement and /or conformations of the molecules in the crystal Lattice. Thus, in the strict sense, polymorphs are different crystalline forms of the same pure substance in which the molecules have different arrangements and / or different configurations of the molecules”. Different polymorphs may differ in their physical properties such as melting point, solubility, X-ray diffraction patterns, etc. Polymorphic forms of a compound can be distinguished in the laboratory by analytical methods such as X-ray diffraction (XRD), Differential Scanning Calorimetry (DSC) and Infrared spectrometry (IR).
Solvent medium and mode of crystallization play very important role in obtaining a crystalline form over the other.
Erlotinib hydrochloride can exist in different polymorphic forms, which differ from each other in terms of stability, physical properties, spectral data and methods of preparation.
The U.S. Patent No. 5,747,498 (herein after referred to as the ‘498 patent) makes no reference to the existence of specific polymorphic forms of erlotinibhydrochloride. In this patent, it is disclosed that the compound is isolated according to conventional techniques; more precisely, according to the embodiments exemplified, crude erlotinib hydrochloride residue (obtained by reaction of 4-chloro-6,7-bis-(2-methoxyethoxy)-quinazoline with 3-ethynylaniline or its hydrochloride salt in a solvent such as a d-Cβ-alcohol, dimethylformamide, N-methylpyrrolidin-2-one, chloroform, acetonitrile, tetrahydrofuran, 1,4-dioxane, pyridine or other aprotic solvents, preferably isopropanol) is basified with saturated aqueous NaHCO3 in the presence of methanol and chloroform followed by flash chromatography on silica using 30% acetone in hexane to afford erlotinib free base, which is further treated with hydrochloric acid in the presence of diethyl ether and chloroform to give erlotinib hydrochloride (melting point: 228° – 2300C).
PCT Patent Publication No. WO 99/55683 disclosed erlotinib mesylate anhydrate and hydrate polymorphic forms, their method of preparation and pharmaceutical compositions containing thereof.
PCT Patent Publication No. WO 01/34574 A1 (herein after referred to as the ‘574 patent publication) described two crystalline forms of erlotinib hydrochloride (polymorph A and polymorph B), characterized by powder X-ray diffraction (p-XRD) pattern. The publication further taught that the synthetic procedure described and exemplified in the ‘498 patent produces the erlotinib hydrochloride as a mixture of the polymorphs A and B.
TARCEVA (erlotinib), a kinase inhibitor, is a quinazolinamine with the chemical name N-(3-ethynylphenyl)-6,7-bis(2-methoxyethoxy)-4-quinazolinamine. TARCEVA contains erlotinib as the hydrochloride salt that has the following structural formula:
 |
Erlotinib hydrochloride has the molecular formula C22H23N3O4•HCl and a molecular weight of 429.90. The molecule has a pKa of 5.42 at 25oC. Erlotinib hydrochloride is very slightly soluble in water, slightly soluble in methanol and practically insoluble in acetonitrile, acetone, ethyl acetate and hexane.
Aqueous solubility of erlotinib hydrochloride is dependent on pH with increased solubility at a pH of less than 5 due to protonation of the secondary amine. Over the pH range of 1.4 to 9.6, maximal solubility of approximately 0.4 mg/mL occurs at a pH of approximately 2.
wo 2012028861
wo2007060691
9
AMUVATINIB










AMUVATINIB
| Name | N-(3,4-Methylenedioxiphenylmethyl) -4 – (benzofuro [3,2-d] pyrimidin-4-yl) piperazine-1-carbothioamide. |
| CAS | 850879-09-3 |
| Formula | C 23 H 21 N 5 O 3 S |
| MW | 447.51 |
| Synonim | MN-470, SGI-0470-03 |
Amuvatinib (MP-470) is an orally bioavailable synthetic carbothioamide with potential antineoplastic activity. Multitargeted receptor tyrosine kinase inhibitor MP470 binds to mutant forms of the stem cell factor receptor (c-Kit; SCFR), inhibiting clinically relevant mutants of this receptor tyrosine kinase that may be associated with resistance to therapy. In addition, MP470 inhibits activities of other receptor tyrosine kinases, such as c-Met, Ret oncoprotein, and mutant forms of Flt3 and PDGFR alpha, which are frequently dysregulated in variety of tumors. This agent also suppresses the induction of DNA repair protein Rad51, thereby potentiating the activities of DNA damage-inducing agents. Mutant forms of c-Kit are often associated with tumor chemoresistance.
Scheme 1

EXAMPLE 34 Synthesis and Analysis of Further Illustrative Compounds Compound (111-1-3), also referred to herein as HPK56/MP-470, is an illustrative compound of the present invention having the following structure:

Analogues of (111-1 -3) were designed and synthesized in order to evaluate and optimize kinase selectivity, aqueous solubility, and to improve pharmacokinetic and pharmacodynamic profiles. Illustrative synthesis approaches for generating (111-1 -3) analogues are depicted in the synthesis schemes below. Synthesis of R-i substituted benzofuranopyrimidines was undertaken. The methyl 3-guanidinobenzofuran-2-carboxylate is prepared from methyl 3-aminobenzofuran-2-carboxylate by reacting with cyanoacetamide in presence of dioxane and dry HCI gas. The obtained guanidine is cyclized in the presence of aqueous NaOH. Similar procedures were utilized for preparing 2- substituted (111-1-3) and its analogues as depicted in the Schemes 8-10 set forth below. Introduction of -NH2 at the 2 position was utilized for various sulfonic, inorganic and hydroxyacid salts. Illustrative compounds are shown in Table 4 below.Table 4


Scheme 1

Scheme 2 Scheme 3
,-NH2 /N Cl S

EXAMPLE 35 Analysis of Compound Binding and Inhibitory Activity against c-kit Mutants
The published crystal structure of c-kit kinase (pdb code:1 PKG) and its mutated structure were used to study the mode of binding of compound (111-1-3) (HPK56/MP-470), a benzofuranopyrimidine compound, its 2-substituted analogs, and quinazoline derivatives.

(Ill- 1-3)
Information about this agent
|
According to news published on 15 Apr 2008; Research data build upon previous results showing that MP-470 exhibits anti-tumor activity in breast and http://www.medicalnewstoday.com/articles/150086.php”>prostate cancer cells. The fact that MP-470 in combination with erlotinib effectively suppressed the HER pathway suggests that concurrent administration of both compounds could represent a new treatment for prostate and breast cancers.
References
|
1. Kreidberg, Jordan A.; Qin, Shan. Method using a cMET inhibitor for the treatment of polycystic kidney disease. PCT Int. Appl. (2009), 43pp. CODEN: PIXXD2 WO 2009111529 A2 20090911 CAN 151:350826 AN 2009:1107388
2. Fujiwara, Masahiro; Fujita, Masayuki. Curing agents, adhesive curable compositions, articles and coating materials therefrom, and optical materials formed by using the compositions. Jpn. Kokai Tokkyo Koho (2008), 39pp. CODEN: JKXXAF JP 2008260894 A 20081030 CAN 149:472741 AN 2008:1303932
3. Janne, Pasi A.; Engelman, Jeffrey; Cantley, Lewis C. Methods for treating cancer resistant to ErbB therapeutics. PCT Int. Appl. (2008), 96pp. CODEN: PIXXD2 WO 2008127710 A2 20081023 CAN 149:486836 AN 2008:1282443
4. Nakagawa, Kiyoshi; Kurushima, Yoshiaki; Mizuma, Masahiro. Fabric with highly-expanded layer and process for production thereof. PCT Int. Appl. (2007), 44pp. CODEN: PIXXD2 WO 2007083641 A1 20070726 CAN 147:213021 AN 2007:815020
5. Mahadevan, D.; Cooke, L.; Riley, C.; Swart, R.; Simons, B.; Della Croce, K.; Wisner, L.; Iorio, M.; Shakalya, K.; Garewal, H.; Nagle, R.; Bearss, D. A novel tyrosine kinase switch is a mechanism of imatinib resistance in gastrointestinal stromal tumors. Oncogene (2007), 26(27), 3909-3919. CODEN: ONCNES ISSN:0950-9232. CAN 147:226484 AN 2007:613908
6. Gong, QingJie; Han, DongYu; Wang, YuRong. Experimental determination of scheelite solubility in 4.0% NaCl solution in critical region. Yanshi Xuebao (2006), 22(12), 3052-3058. CODEN: YANXEU ISSN:1000-0569. CAN 147:34728 AN 2007:301330
7. Tokunaga, Koji; Mukai, Takashi; Kishigami, Akira. Two-component weak solvent-based coating compositions with no curing interference by dewing. Jpn. Kokai Tokkyo Koho (2005), 15 pp. CODEN: JKXXAF JP 2005239815 A 20050908 CAN 143:249850 AN 2005:979155
8. Lauer, S. J.; Shuster, J. J.; Mahoney, D. H., Jr.; Winick, N.; Toledano, S.; Munoz, L.; Kiefer, G.; Pullen, J. D.; Steuber, C. P.; Camitta, B. M. A comparison of early intensive methotrexate/mercaptopurine with early intensive alternating combination chemotherapy for high-risk B-precursor acute lymphoblastic leukemia: A pediatric oncology group phase III randomized trial. Leukemia (2001), 15(7), 1038-1045. CODEN: LEUKED ISSN:0887-6924. CAN 135:338846 AN 2001:586045
9. Sen, Ayusman; Hennis, April. Palladium (II) catalyzed polymerization of norbornene and acrylates. PCT Int. Appl. (2001), 22 pp. CODEN: PIXXD2 WO 2001021670 A1 20010329 CAN 134:252774 AN 2001:228935
10. Patel, Raman; Mallin, Dan; Saunders, Keith; Tiberio, Patrick; Andries, John. Polymer compositions containing polyolefins, polar polymers and block or graft copolymer compatibilizers and their preparation. PCT Int. Appl. (1999), 25 pp. CODEN: PIXXD2 WO 9950350 A1 19991007 CAN 131:272658 AN 1999:640932
11. Koehler, Burkhard; Imai, Seisaku; Doering, Joachim; Ruesseler, Wolfgang; Dorf, Ernst Ullrich. Mixtures of polyarylene sulfides, glass fibers, and polymaleimides with good mechanical properties. Ger. Offen. (1992), 4 pp. CODEN: GWXXBX DE 4105913 A1 19920827 CAN 118:82122 AN 1993:82122
12. Itoh, Michiya; Fuke, Kiyokazu; Kobayashi, Sachiko. Direct observation of intramolecular anthracene excimer in 1,3-dianthrylpropane. Journal of Chemical Physics (1980), 72(2), 1417-18. CODEN: JCPSA6 ISSN:0021-9606. CAN 92:155340 AN 1980:155340
13. Tomillero A; Moral M A Gateways to clinical trials. Methods and findings in experimental and clinical pharmacology (2009), 31(9), 597-633. Journal code: 7909595. ISSN:0379-0355. PubMed ID 20094643 AN 2010048694
14. Tomillero A; Moral M A Gateways to clinical trials. Methods and findings in experimental and clinical pharmacology (2009), 31(8), 541-57. Journal code: 7909595. ISSN:0379-0355. PubMed ID 19967103 AN 2009808578
15. Gorrand Jean-Marie; Doly Michel; Bacin Franck Macular pigment density assessed by directional fundus reflectance. Journal of the Optical Society of America. A, Optics, image science, and vision (2009), 26(8), 1847-54. Journal code: 9800943. ISSN:1084-7529. PubMed ID 19649122 AN 2009529589
16. Tomillero A; Moral M A Gateways to clinical trials. Methods and findings in experimental and clinical pharmacology (2009), 31(4), 263-98. Journal code: 7909595. ISSN:0379-0355. PubMed ID 19557204 AN 2009445725

ReplyDeletei found your article very intresting. really nice post. keep posting this type of important post. lots of love from rajasthan also help me to improve my backlinking i am adding my website link.
https://onlinepharmacypill.com/product/soma-500-mg/
Soma 500mg
Buy Soma 500mg
Soma 500mg Online
Body pain is one of those health related problems and it is very common that people have to go through various types of pains. Now no
more worries related to these pain problems, as to treat your pain you can buy painkillers online. . Now you can buy Soma 500mg online at affordable price